State Key Laboratory of Systems Medicine for Cancer, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, School of Medicine and School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, China.
School of Biomedical Engineering and Med-X Research Institute, Shanghai Jiao Tong University, Shanghai, China.
Clin Transl Med. 2023 Nov;13(11):e1475. doi: 10.1002/ctm2.1475.
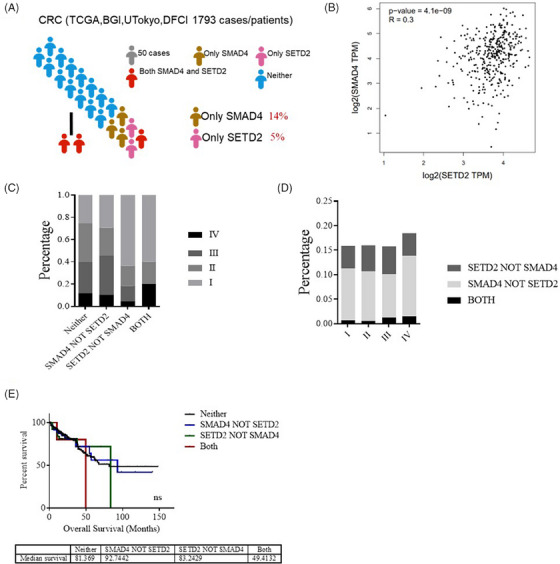
Colorectal cancer (CRC) is a complex, multistep disease that arises from the interplay genetic mutations and epigenetic alterations. The histone H3K36 trimethyltransferase SET domain-containing 2 (SETD2), as an epigenetic signalling molecule, has a 5% mutation rate in CRC. SETD2 expression is decreased in the development of human CRC and mice treated with Azoxymethane /Dextran sodium sulfate (AOM/DSS). Loss of SETD2 promoted CRC development. SMAD Family member 4 (SMAD4) has a 14% mutation rate in CRC, and SMAD4 ablation leads to CRC. The co-mutation of SETD2 and SMAD4 predicted advanced CRC. However, little is known on the potential synergistic effect of SETD2 and SMAD4.
CRC tissues from mice and SW620 cells were used as research subjects. Clinical databases of CRC patients were analyzed to investigate the association between SETD2 and SMAD4. SETD2 and SMAD4 double-knockout mice were established to further investigate the role of SETD2 in SMAD4-deficient CRC. The intestinal epithelial cells (IECs) were isolated for RNA sequencing and chromatin immunoprecipitation sequencing (ChIP-seq) to explore the mechanism and the key molecules resulting in CRC. Molecular and cellular experiments were conducted to analyze the role of SETD2 in SMAD4-deficient CRC. Finally, rescue experiments were performed to confirm the molecular mechanism of SETD2 in the development of SMAD4-dificient CRC.
The deletion of SETD2 promotes the malignant progression of SMAD4-deficient CRC. Smad4 ; Setd2 mice developed a more severe CRC phenotype after AOM/DSS induction, with a larger tumour size and a more vigorous epithelial proliferation rate. Further mechanistic findings revealed that the loss of SETD2 resulted in the down-regulation of DUSP7, which is involved in the inhibition of the RAS/ERK signalling pathway. Finally, the ERK1/2 inhibitor SCH772984 significantly attenuated the progression of CRC in Smad4 ;Setd2 mice, and overexpression of DUSP7 significantly inhibited the proliferation rates of SETD2 ; SMAD4 SW620 cells.
Our results demonstrated that SETD2 inhibits the RAS/ERK signaling pathway by facilitating the transcription of DUSP7 in SMAD4-deficient CRC, which could provide a potential therapeutic target for the treatment of advanced CRC.
结直肠癌(CRC)是一种复杂的多步骤疾病,由遗传突变和表观遗传改变相互作用引起。组蛋白 H3K36 三甲基转移酶 SET 域包含 2 (SETD2)作为一种表观遗传信号分子,在 CRC 中的突变率为 5%。在人类 CRC 的发展和用氧化偶氮甲烷/葡聚糖硫酸钠(AOM/DSS)处理的小鼠中,SETD2 的表达减少。SETD2 的缺失促进了 CRC 的发展。SMAD 家族成员 4(SMAD4)在 CRC 中的突变率为 14%,SMAD4 的缺失导致 CRC。SETD2 和 SMAD4 的共突变预测 CRC 进展。然而,关于 SETD2 和 SMAD4 之间潜在协同作用的知之甚少。
以小鼠 CRC 组织和 SW620 细胞为研究对象。分析 CRC 患者的临床数据库,以研究 SETD2 和 SMAD4 之间的关系。建立 SETD2 和 SMAD4 双重敲除小鼠,进一步研究 SETD2 在 SMAD4 缺陷型 CRC 中的作用。分离肠上皮细胞(IECs)进行 RNA 测序和染色质免疫沉淀测序(ChIP-seq),以探讨导致 CRC 的机制和关键分子。进行分子和细胞实验,以分析 SETD2 在 SMAD4 缺陷型 CRC 中的作用。最后,进行挽救实验以确认 SETD2 在 SMAD4 缺陷型 CRC 发展中的分子机制。
SETD2 的缺失促进了 SMAD4 缺陷型 CRC 的恶性进展。AOM/DSS 诱导后,Smad4;Setd2 小鼠发展出更严重的 CRC 表型,肿瘤体积更大,上皮增殖率更高。进一步的机制研究发现,SETD2 的缺失导致 DUSP7 的下调,DUSP7 参与抑制 RAS/ERK 信号通路。最后,ERK1/2 抑制剂 SCH772984 显著减轻 Smad4;Setd2 小鼠 CRC 的进展,过表达 DUSP7 显著抑制 SETD2;SMAD4 SW620 细胞的增殖率。
我们的研究结果表明,SETD2 通过促进 DUSP7 在 SMAD4 缺陷型 CRC 中的转录来抑制 RAS/ERK 信号通路,这为治疗晚期 CRC 提供了一个潜在的治疗靶点。