Yu Shirui, Wang Ziyang, Nan Jiale, Li Aihua, Yang Xuemei, Tang Xiaoli
Institute of Medical Information, Chinese Academy of Medical Sciences, Beijing, China.
JMIR Form Res. 2023 Nov 15;7:e50998. doi: 10.2196/50998.
Schizophrenia is a serious mental disease. With increased research funding for this disease, schizophrenia has become one of the key areas of focus in the medical field. Searching for associations between diseases and genes is an effective approach to study complex diseases, which may enhance research on schizophrenia pathology and lead to the identification of new treatment targets.
The aim of this study was to identify potential schizophrenia risk genes by employing machine learning methods to extract topological characteristics of proteins and their functional roles in a protein-protein interaction (PPI)-keywords (PPIK) network and understand the complex disease-causing property. Consequently, a PPIK-based metagraph representation approach is proposed.
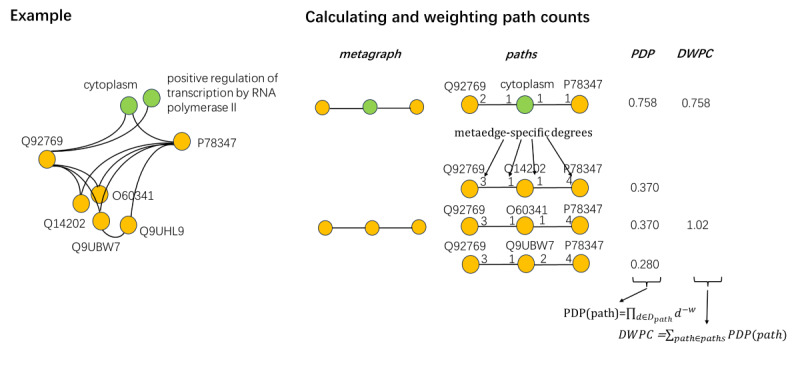
To enrich the PPI network, we integrated keywords describing protein properties and constructed a PPIK network. We extracted features that describe the topology of this network through metagraphs. We further transformed these metagraphs into vectors and represented proteins with a series of vectors. We then trained and optimized our model using random forest (RF), extreme gradient boosting, light gradient boosting machine, and logistic regression models.
Comprehensive experiments demonstrated the good performance of our proposed method with an area under the receiver operating characteristic curve (AUC) value between 0.72 and 0.76. Our model also outperformed baseline methods for overall disease protein prediction, including the random walk with restart, average commute time, and Katz models. Compared with the PPI network constructed from the baseline models, complementation of keywords in the PPIK network improved the performance (AUC) by 0.08 on average, and the metagraph-based method improved the AUC by 0.30 on average compared with that of the baseline methods. According to the comprehensive performance of the four models, RF was selected as the best model for disease protein prediction, with precision, recall, F1-score, and AUC values of 0.76, 0.73, 0.72, and 0.76, respectively. We transformed these proteins to their encoding gene IDs and identified the top 20 genes as the most probable schizophrenia-risk genes, including the EYA3, CNTN4, HSPA8, LRRK2, and AFP genes. We further validated these outcomes against metagraph features and evidence from the literature, performed a features analysis, and exploited evidence from the literature to interpret the correlation between the predicted genes and diseases.
The metagraph representation based on the PPIK network framework was found to be effective for potential schizophrenia risk genes identification. The results are quite reliable as evidence can be found in the literature to support our prediction. Our approach can provide more biological insights into the pathogenesis of schizophrenia.

精神分裂症是一种严重的精神疾病。随着对该疾病研究资金的增加,精神分裂症已成为医学领域的关键关注领域之一。寻找疾病与基因之间的关联是研究复杂疾病的有效方法,这可能会加强对精神分裂症病理学的研究,并有助于确定新的治疗靶点。
本研究旨在通过运用机器学习方法提取蛋白质的拓扑特征及其在蛋白质-蛋白质相互作用(PPI)-关键词(PPIK)网络中的功能作用,识别潜在的精神分裂症风险基因,并了解复杂的致病特性。因此,提出了一种基于PPIK的元图表示方法。
为了丰富PPI网络,我们整合了描述蛋白质特性的关键词并构建了PPIK网络。我们通过元图提取了描述该网络拓扑结构的特征。我们进一步将这些元图转换为向量,并用一系列向量表示蛋白质。然后,我们使用随机森林(RF)、极端梯度提升、轻梯度提升机和逻辑回归模型对模型进行训练和优化。
综合实验表明,我们提出的方法具有良好的性能,受试者工作特征曲线(AUC)值在0.72至0.76之间。我们的模型在整体疾病蛋白质预测方面也优于基线方法,包括重启随机游走、平均通勤时间和Katz模型。与从基线模型构建的PPI网络相比,PPIK网络中关键词的补充平均将性能(AUC)提高了0.08,基于元图的方法与基线方法相比平均将AUC提高了0.30。根据四个模型的综合性能,RF被选为疾病蛋白质预测的最佳模型,其精确率、召回率、F1分数和AUC值分别为0.76、0.73、0.72和0.76。我们将这些蛋白质转换为它们的编码基因ID,并将前20个基因确定为最有可能的精神分裂症风险基因,包括EYA3、CNTN4、HSPA8、LRRK2和AFP基因。我们进一步根据元图特征和文献证据验证了这些结果,进行了特征分析,并利用文献证据解释了预测基因与疾病之间的相关性。
基于PPIK网络框架的元图表示被发现对识别潜在的精神分裂症风险基因有效。结果相当可靠,因为文献中可以找到支持我们预测的证据。我们的方法可以为精神分裂症的发病机制提供更多生物学见解。