College of Information Engineering, Zhejiang University of Technology.
researcher of AI in the BioMap.
Brief Bioinform. 2023 Nov 22;25(1). doi: 10.1093/bib/bbad420.
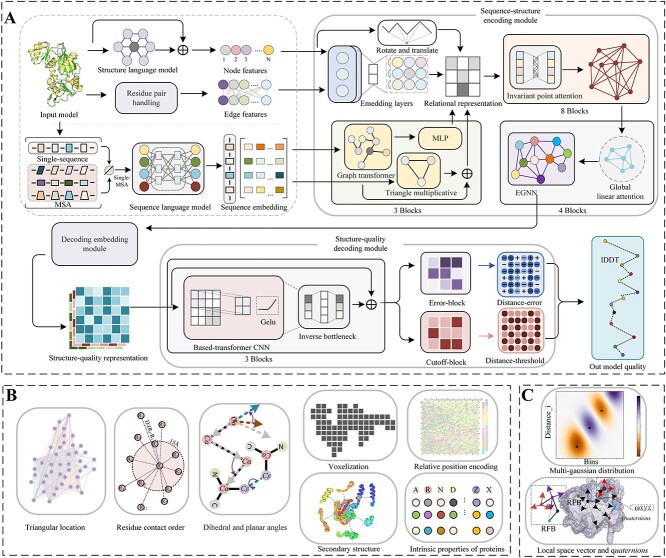
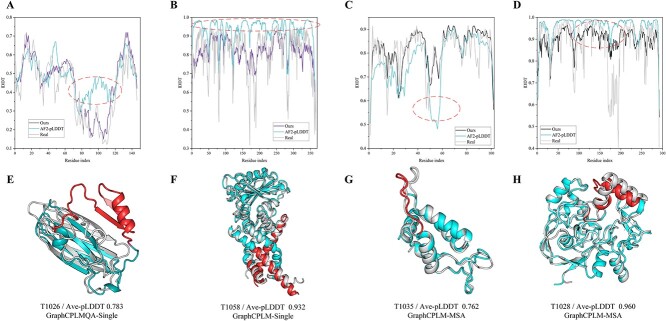
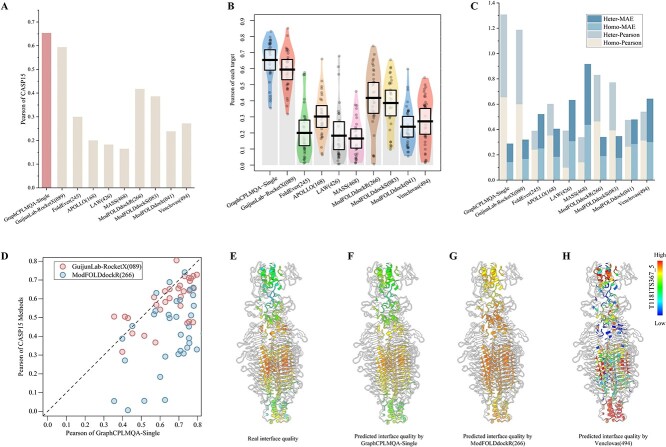
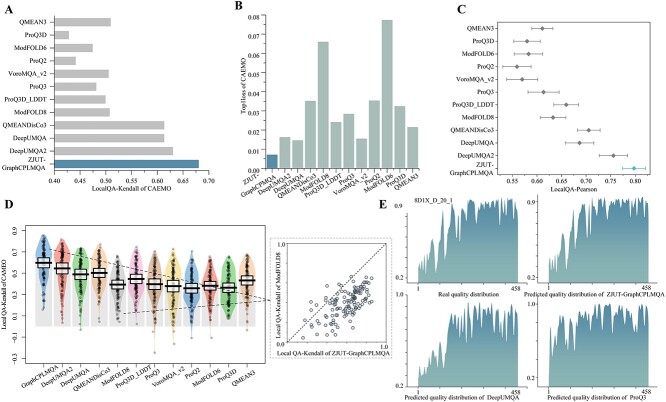
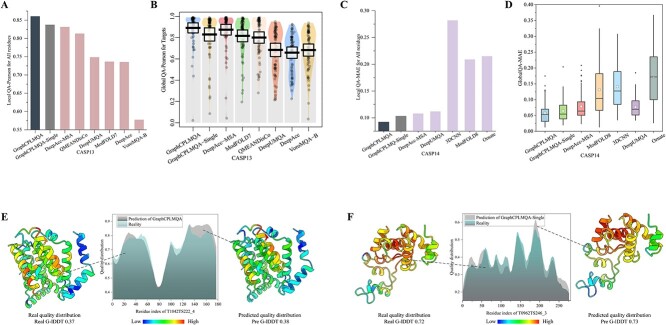
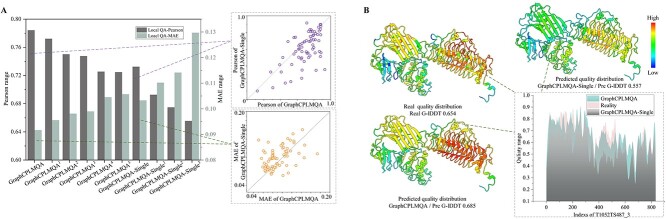
Model quality evaluation is a crucial part of protein structural biology. How to distinguish high-quality models from low-quality models, and to assess which high-quality models have relatively incorrect regions for improvement, are remain a challenge. More importantly, the quality assessment of multimer models is a hot topic for structure prediction. In this study, we propose GraphCPLMQA, a novel approach for evaluating residue-level model quality that combines graph coupled networks and embeddings from protein language models. The GraphCPLMQA consists of a graph encoding module and a transform-based convolutional decoding module. In encoding module, the underlying relational representations of sequence and high-dimensional geometry structure are extracted by protein language models with Evolutionary Scale Modeling. In decoding module, the mapping connection between structure and quality is inferred by the representations and low-dimensional features. Specifically, the triangular location and residue level contact order features are designed to enhance the association between the local structure and the overall topology. Experimental results demonstrate that GraphCPLMQA using single-sequence embedding achieves the best performance compared with the CASP15 residue-level interface evaluation methods among 9108 models in the local residue interface test set of CASP15 multimers. In CAMEO blind test (20 May 2022 to 13 August 2022), GraphCPLMQA ranked first compared with other servers (https://www.cameo3d.org/quality-estimation). GraphCPLMQA also outperforms state-of-the-art methods on 19, 035 models in CASP13 and CASP14 monomer test set.
模型质量评估是蛋白质结构生物学的一个重要组成部分。如何区分高质量模型和低质量模型,以及评估哪些高质量模型具有需要改进的相对错误区域,仍然是一个挑战。更重要的是,多聚体模型的质量评估是结构预测的一个热门话题。在这项研究中,我们提出了 GraphCPLMQA,这是一种结合了图耦合网络和蛋白质语言模型的嵌入的残基水平模型质量评估的新方法。GraphCPLMQA 由图编码模块和基于变换的卷积解码模块组成。在编码模块中,通过进化尺度建模的蛋白质语言模型提取序列和高维几何结构的基本关系表示。在解码模块中,通过表示和低维特征推断结构和质量之间的映射关系。具体来说,设计了三角形位置和残基级接触顺序特征来增强局部结构与整体拓扑之间的关联。实验结果表明,与 CASP15 多聚体局部残基界面测试集中的 9108 个模型中的其他 CASP15 残基界面评估方法相比,GraphCPLMQA 使用单序列嵌入实现了最佳性能。在 CAMEO 盲测(2022 年 5 月 20 日至 2022 年 8 月 13 日)中,GraphCPLMQA 与其他服务器(https://www.cameo3d.org/quality-estimation)相比排名第一。GraphCPLMQA 在 CASP13 和 CASP14 单体测试集中的 19035 个模型上的表现也优于最先进的方法。