Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI, 48109, USA.
Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Ann Arbor, MI, 48109, USA.
Genome Biol. 2023 Dec 21;24(1):294. doi: 10.1186/s13059-023-03144-z.
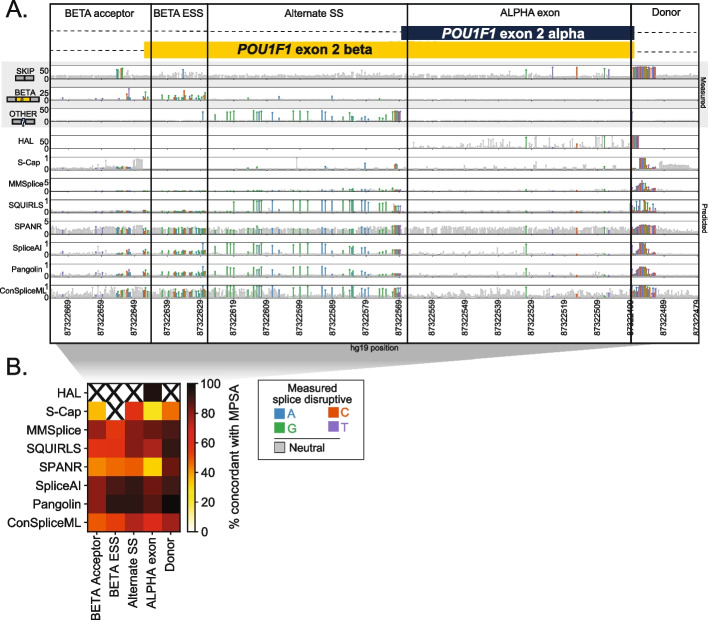
Variants that disrupt mRNA splicing account for a sizable fraction of the pathogenic burden in many genetic disorders, but identifying splice-disruptive variants (SDVs) beyond the essential splice site dinucleotides remains difficult. Computational predictors are often discordant, compounding the challenge of variant interpretation. Because they are primarily validated using clinical variant sets heavily biased to known canonical splice site mutations, it remains unclear how well their performance generalizes.
We benchmark eight widely used splicing effect prediction algorithms, leveraging massively parallel splicing assays (MPSAs) as a source of experimentally determined ground-truth. MPSAs simultaneously assay many variants to nominate candidate SDVs. We compare experimentally measured splicing outcomes with bioinformatic predictions for 3,616 variants in five genes. Algorithms' concordance with MPSA measurements, and with each other, is lower for exonic than intronic variants, underscoring the difficulty of identifying missense or synonymous SDVs. Deep learning-based predictors trained on gene model annotations achieve the best overall performance at distinguishing disruptive and neutral variants, and controlling for overall call rate genome-wide, SpliceAI and Pangolin have superior sensitivity. Finally, our results highlight two practical considerations when scoring variants genome-wide: finding an optimal score cutoff, and the substantial variability introduced by differences in gene model annotation, and we suggest strategies for optimal splice effect prediction in the face of these issues.
SpliceAI and Pangolin show the best overall performance among predictors tested, however, improvements in splice effect prediction are still needed especially within exons.
在许多遗传疾病中,破坏 mRNA 剪接的变异体占致病负担的相当大一部分,但识别超出必需剪接位点二核苷酸的剪接破坏变异体(SDV)仍然很困难。计算预测器通常存在差异,这增加了变异解释的挑战。由于它们主要使用偏向已知经典剪接位点突变的临床变异集进行验证,因此尚不清楚它们的性能如何普遍适用。
我们利用大规模并行剪接分析(MPSA)作为实验确定的真实值来源,对 8 种广泛使用的剪接效应预测算法进行基准测试。MPSA 同时检测许多变体,以提名候选 SDV。我们将 5 个基因中 3616 个变体的实验测量的剪接结果与生物信息学预测进行比较。对于外显子变体,算法与 MPSA 测量值的一致性以及彼此之间的一致性低于内含子变体,这突出了识别错义或同义 SDV 的困难。基于深度学习的基于基因模型注释的预测器在区分破坏和中性变体方面表现出最佳的整体性能,并且在全基因组范围内控制总体调用率方面,SpliceAI 和 Pangolin 具有更高的灵敏度。最后,我们的结果突出了在全基因组范围内评分变体时需要考虑的两个实际问题:找到最佳的评分截止值,以及基因模型注释差异引入的大量可变性,我们为在这些问题面前进行最佳剪接效应预测提供了策略。
在测试的预测器中,SpliceAI 和 Pangolin 显示出最佳的整体性能,然而,特别是在内含子中,仍然需要改进剪接效应预测。