VIB-UGent Center for Medical Biotechnology, VIB, Ghent, Belgium; Department of Biomolecular Medicine, Ghent University, Ghent, Belgium; Department of Applied Mathematics, Computer Science and Statistics, Ghent University, Ghent, Belgium.
Laboratoire de Spectrométrie de Masse BioOrganique, IPHC UMR 7178, CNRS, Infrastructure Nationale de Protéomique ProFI - FR2048, Université de Strasbourg, Strasbourg, France.
Mol Cell Proteomics. 2024 Feb;23(2):100708. doi: 10.1016/j.mcpro.2023.100708. Epub 2023 Dec 26.
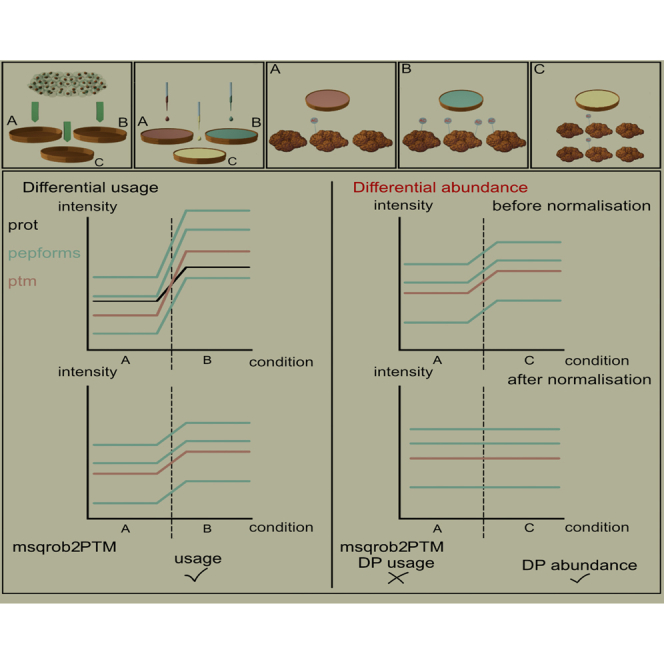
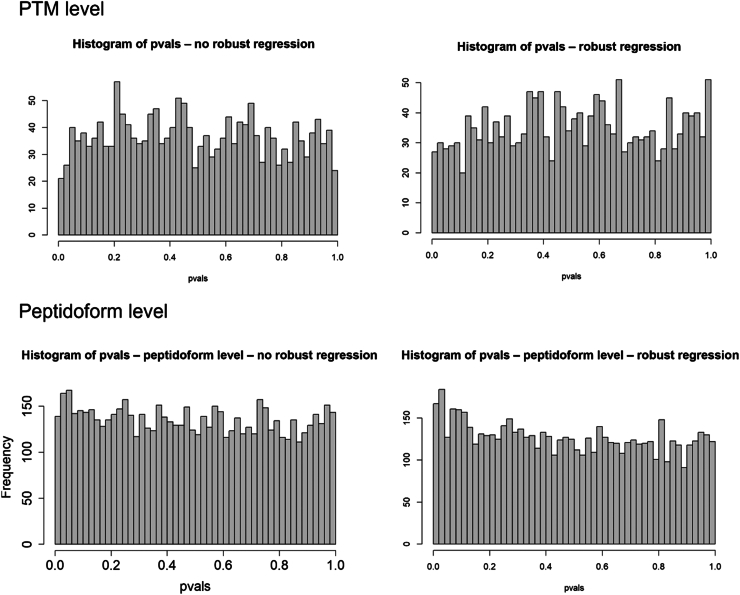
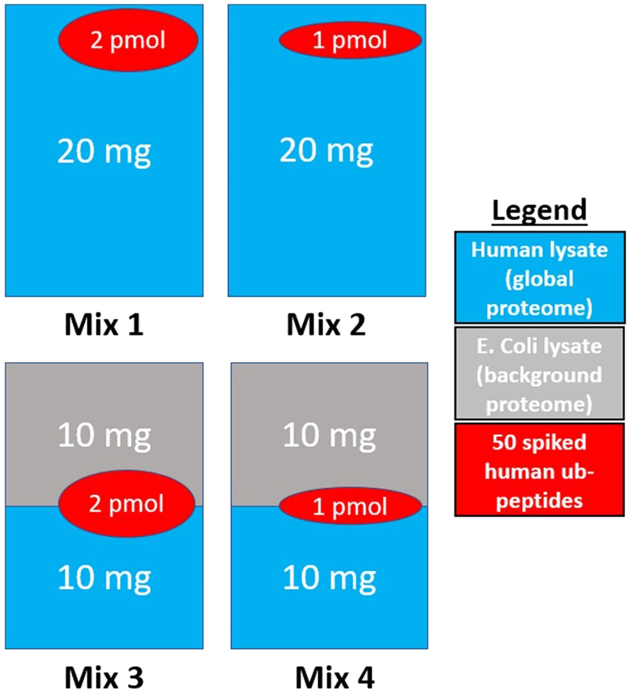
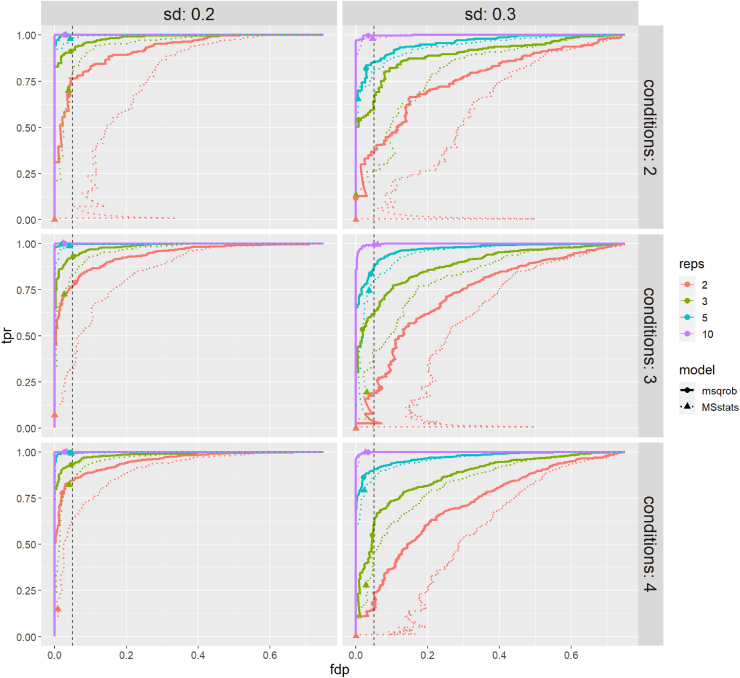
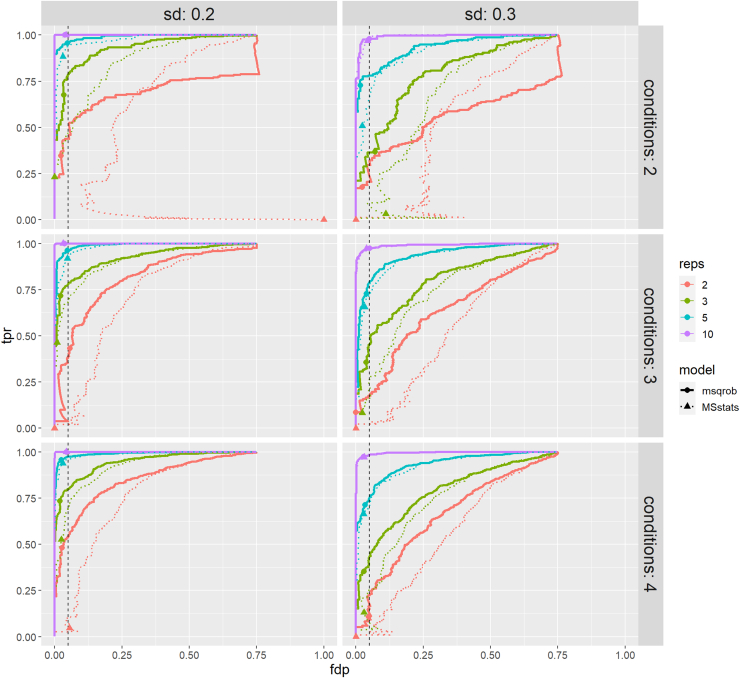
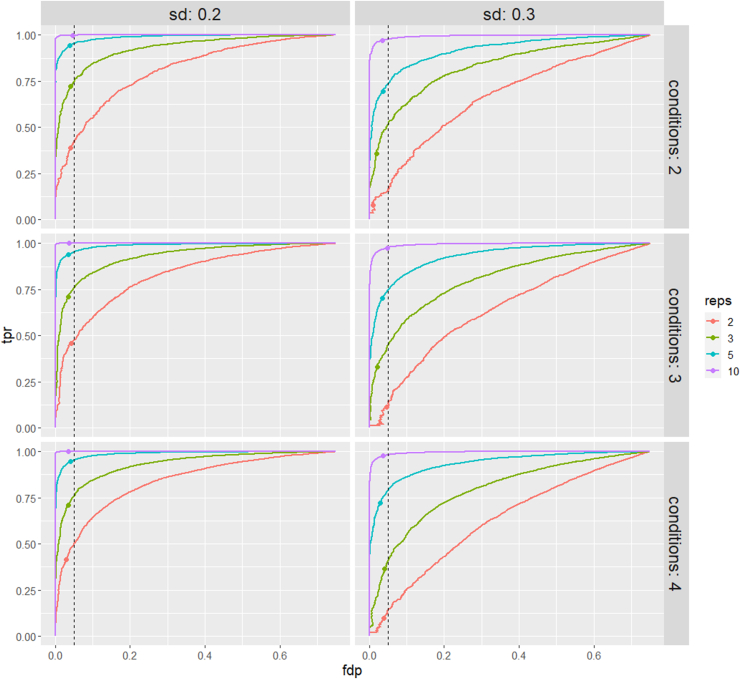
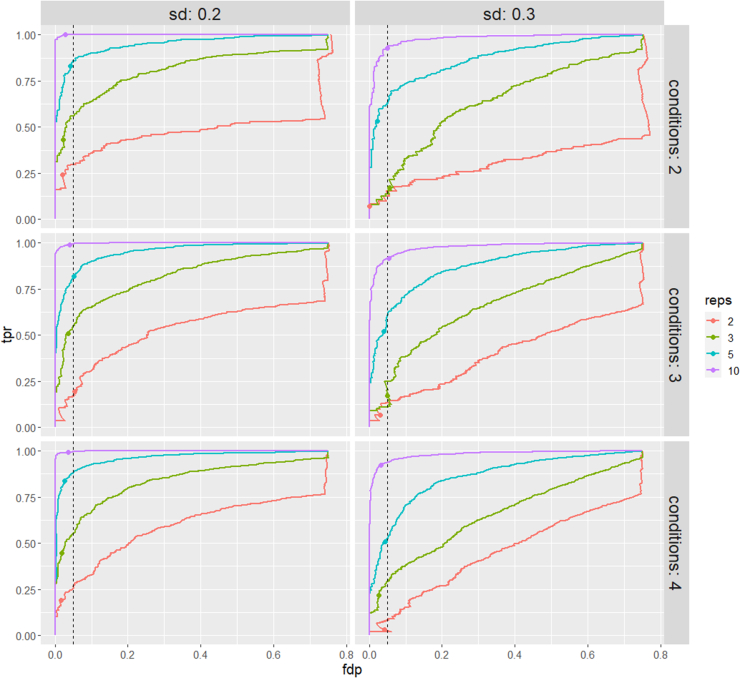
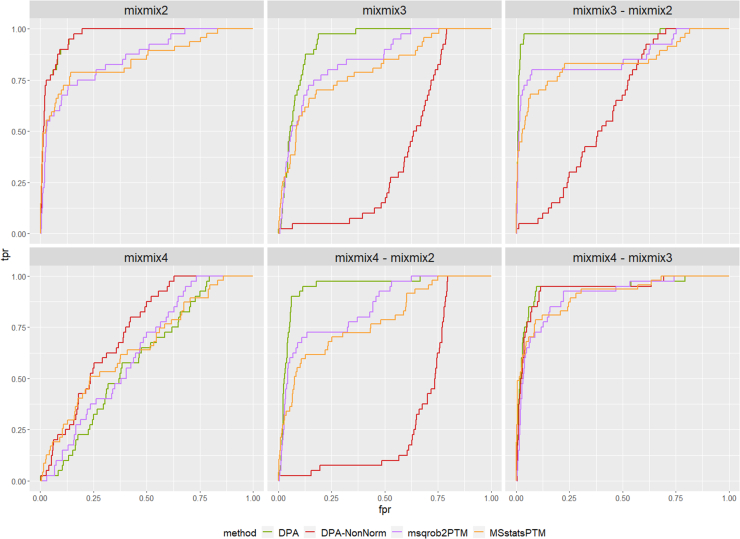
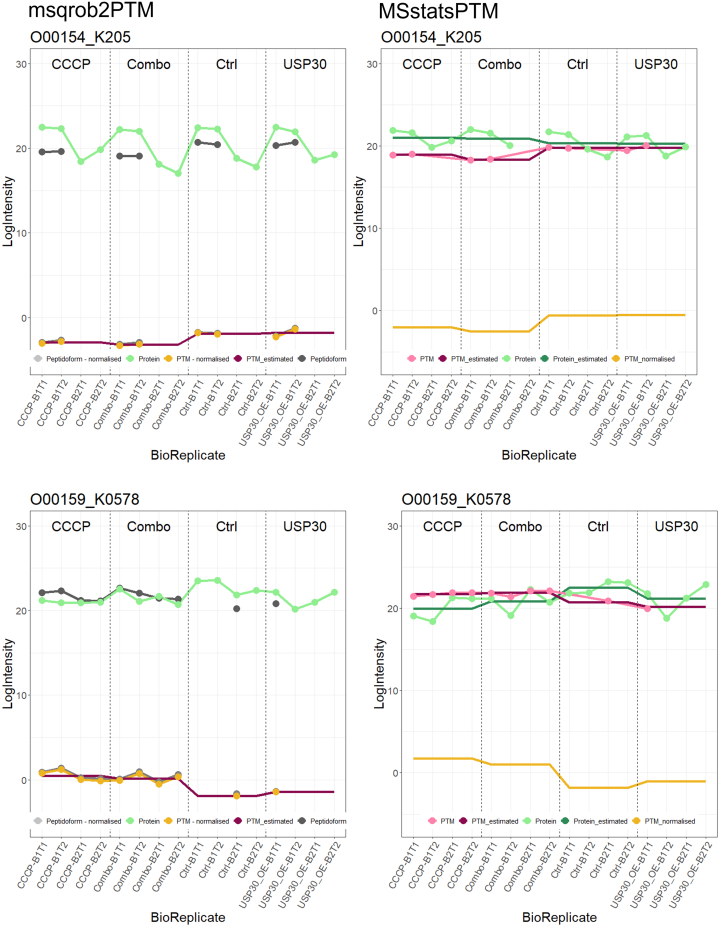
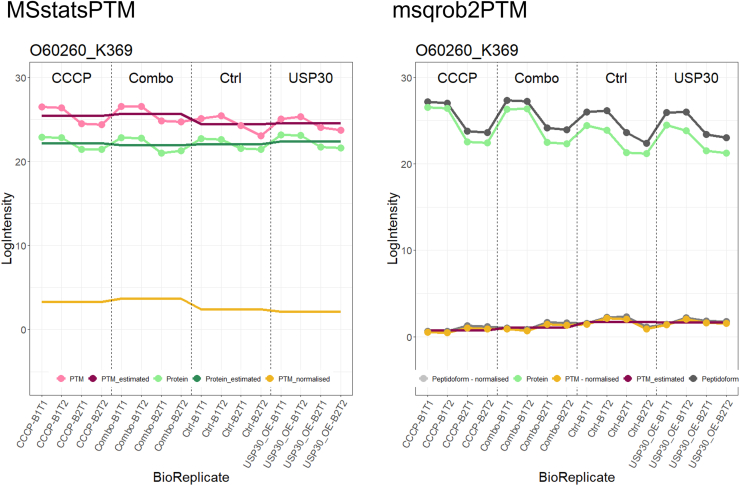
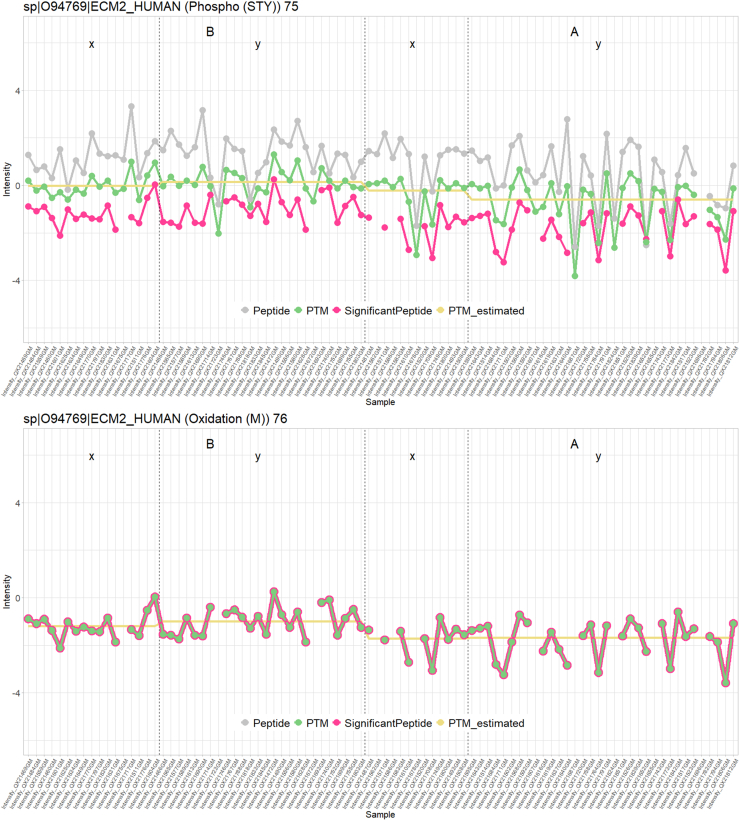
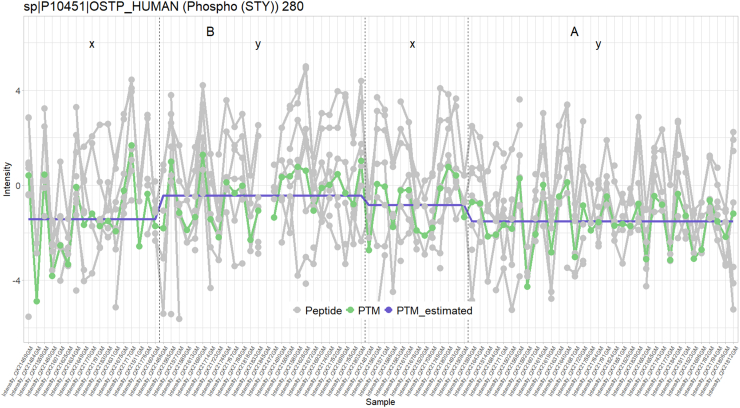
In the era of open-modification search engines, more posttranslational modifications than ever can be detected by LC-MS/MS-based proteomics. This development can switch proteomics research into a higher gear, as PTMs are key in many cellular pathways important in cell proliferation, migration, metastasis, and aging. However, despite these advances in modification identification, statistical methods for PTM-level quantification and differential analysis have yet to catch up. This absence can partly be explained by statistical challenges inherent to the data, such as the confounding of PTM intensities with its parent protein abundance. Therefore, we have developed msqrob2PTM, a new workflow in the msqrob2 universe capable of differential abundance analysis at the PTM and at the peptidoform level. The latter is important for validating PTMs found as significantly differential. Indeed, as our method can deal with multiple PTMs per peptidoform, there is a possibility that significant PTMs stem from one significant peptidoform carrying another PTM, hinting that it might be the other PTM driving the perceived differential abundance. Our workflows can flag both differential peptidoform abundance (DPA) and differential peptidoform usage (DPU). This enables a distinction between direct assessment of differential abundance of peptidoforms (DPA) and differences in the relative usage of peptidoforms corrected for corresponding protein abundances (DPU). For DPA, we directly model the log2-transformed peptidoform intensities, while for DPU, we correct for parent protein abundance by an intermediate normalization step which calculates the log2-ratio of the peptidoform intensities to their summarized parent protein intensities. We demonstrated the utility and performance of msqrob2PTM by applying it to datasets with known ground truth, as well as to biological PTM-rich datasets. Our results show that msqrob2PTM is on par with, or surpassing the performance of, the current state-of-the-art methods. Moreover, msqrob2PTM is currently unique in providing output at the peptidoform level.
在开放修饰搜索引擎时代,基于 LC-MS/MS 的蛋白质组学可以检测到比以往更多的翻译后修饰。这一发展可以将蛋白质组学研究推向更高的水平,因为翻译后修饰是许多对细胞增殖、迁移、转移和衰老至关重要的细胞途径中的关键因素。然而,尽管在修饰鉴定方面取得了这些进展,但翻译后修饰水平定量和差异分析的统计方法仍有待提高。这种缺失部分可以解释为数据固有的统计挑战,例如翻译后修饰强度与其母体蛋白丰度的混淆。因此,我们开发了 msqrob2PTM,这是 msqrob2 宇宙中的一个新工作流程,能够在翻译后修饰和肽形式水平上进行差异丰度分析。后者对于验证作为显著差异的翻译后修饰非常重要。事实上,由于我们的方法可以处理每个肽形式的多个翻译后修饰,因此有一个可能性,即显著的翻译后修饰源自一个携带另一个翻译后修饰的显著肽形式,暗示可能是另一个翻译后修饰驱动了感知到的差异丰度。我们的工作流程可以标记差异肽形式丰度(DPA)和差异肽形式使用(DPU)。这可以区分肽形式直接评估的差异丰度(DPA)和校正相应蛋白质丰度的肽形式相对使用差异(DPU)。对于 DPA,我们直接对 log2 转换后的肽形式强度进行建模,而对于 DPU,我们通过中间标准化步骤校正母体蛋白丰度,该步骤计算肽形式强度与其汇总母体蛋白强度的 log2 比值。我们通过将其应用于具有已知真实值的数据集以及富含生物学翻译后修饰的数据集,证明了 msqrob2PTM 的实用性和性能。我们的结果表明,msqrob2PTM 的性能与当前最先进的方法相当,甚至超过了这些方法。此外,msqrob2PTM 目前是唯一在肽形式水平上提供输出的方法。