Department of Ophthalmology, Centro Hospitalar e Universitário de Coimbra (CHUC), Coimbra, Portugal.
Instituto de Oftalmologia Dr. Gama Pinto (IOGP), Lisboa, Portugal.
Graefes Arch Clin Exp Ophthalmol. 2024 Jun;262(6):1883-1897. doi: 10.1007/s00417-023-06360-2. Epub 2024 Jan 8.
Retinitis pigmentosa (RP) comprises a genetically and clinically heterogeneous group of inherited retinal degenerations, where 20-30% of patients exhibit extra-ocular manifestations (syndromic RP). Understanding the genetic profile of RP has important implications for disease prognosis and genetic counseling. This study aimed to characterize the genetic profile of syndromic RP in Portugal.
Multicenter, retrospective cohort study. Six Portuguese healthcare providers identified patients with a clinical diagnosis of syndromic RP and available genetic testing results. All patients had been previously subjected to a detailed ophthalmologic examination and clinically oriented genetic testing. Genetic variants were classified according to the American College of Medical Genetics and Genomics; only likely pathogenic or pathogenic variants were considered relevant for disease etiology.
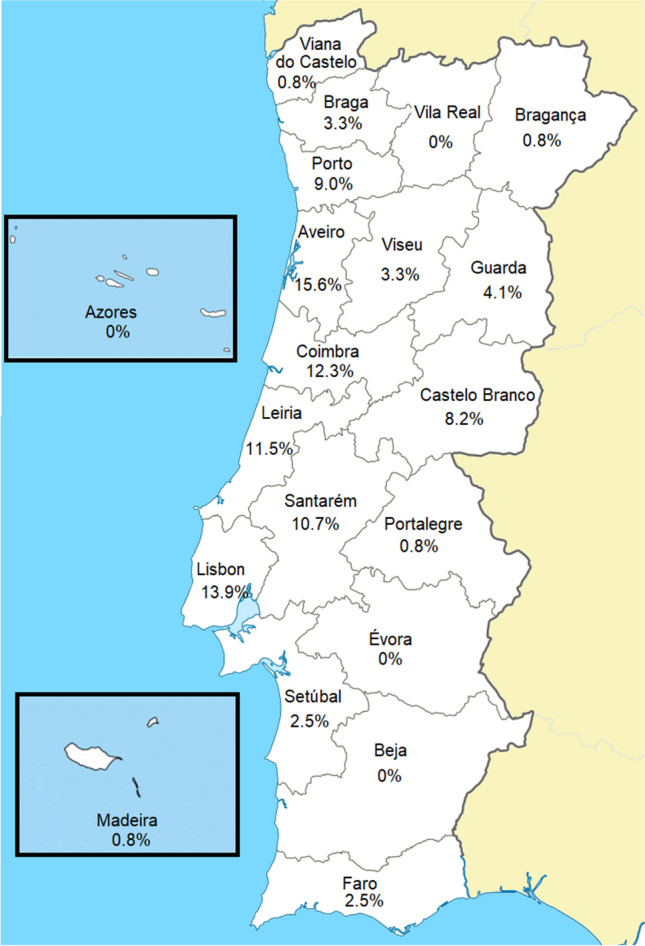
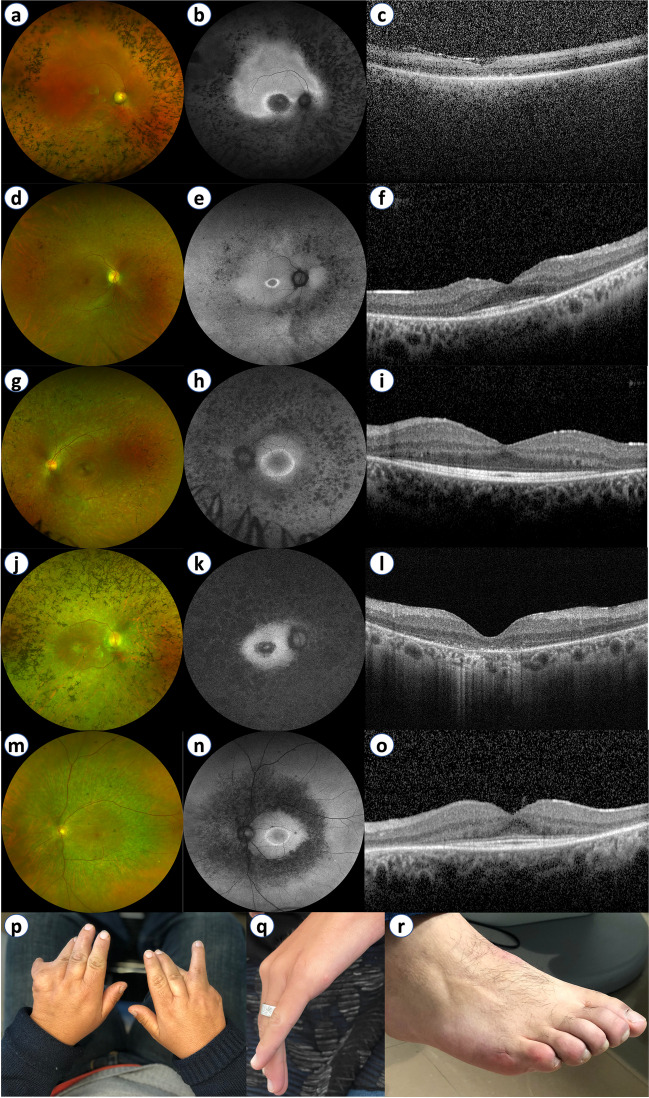
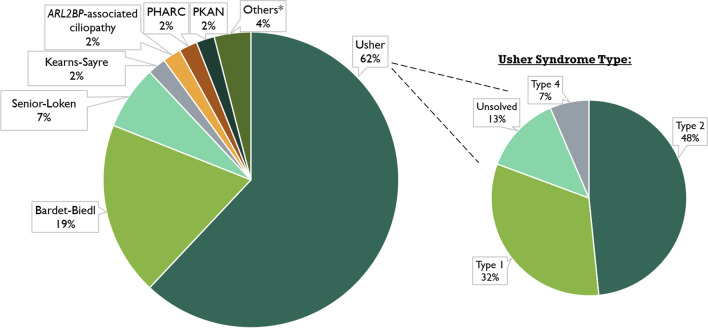
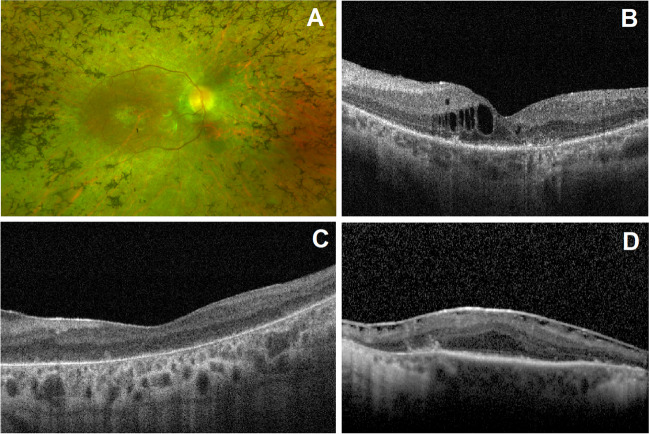
One hundred and twenty-two patients (53.3% males) from 100 families were included. Usher syndrome was the most frequent diagnosis (62.0%), followed by Bardet-Biedl (19.0%) and Senior-Løken syndromes (7.0%). Deleterious variants were identified in 86/100 families for a diagnostic yield of 86.0% (87.1% for Usher and 94.7% for Bardet-Biedl). A total of 81 genetic variants were identified in 25 different genes, 22 of which are novel. USH2A and MYO7A were responsible for most type II and type I Usher syndrome cases, respectively. BBS1 variants were the cause of Bardet-Biedl syndrome in 52.6% of families. Best-corrected visual acuity (BCVA) records were available at baseline and last visit for 99 patients (198 eyes), with a median follow-up of 62.0 months. The mean BCVA was 56.5 ETDRS letters at baseline (Snellen equivalent ~ 20/80), declining to 44.9 ETDRS letters (Snellen equivalent ~ 20/125) at the last available follow-up (p < 0.001).
This is the first multicenter study depicting the genetic profile of syndromic RP in Portugal, thus contributing toward a better understanding of this heterogeneous disease group. Usher and Bardet-Biedl syndromes were found to be the most common types of syndromic RP in this large Portuguese cohort. A high diagnostic yield was obtained, highlighting current genetic testing capabilities in providing a molecular diagnosis to most affected individuals. This has major implications in determining disease-related prognosis and providing targeted genetic counseling for syndromic RP patients in Portugal.
色素性视网膜炎(RP)是一组具有遗传异质性的遗传性视网膜变性,其中 20-30%的患者存在眼外表现(综合征性 RP)。了解 RP 的遗传特征对疾病预后和遗传咨询具有重要意义。本研究旨在描述葡萄牙综合征性 RP 的遗传特征。
多中心回顾性队列研究。六名葡萄牙医疗保健提供者鉴定了具有综合征性 RP 临床诊断和可用基因检测结果的患者。所有患者均接受了详细的眼科检查和临床导向的基因检测。根据美国医学遗传学与基因组学学院的标准对遗传变异进行分类;仅考虑可能致病或致病的变异与疾病病因有关。
共纳入 100 个家庭的 122 名患者(53.3%为男性)。最常见的诊断是 Usher 综合征(62.0%),其次是 Bardet-Biedl 综合征(19.0%)和 Senior-Løken 综合征(7.0%)。在 100 个家庭中,有 86 个家庭发现了有害变异,诊断率为 86.0%(Usher 为 87.1%,Bardet-Biedl 为 94.7%)。在 25 个不同基因中发现了 81 个遗传变异,其中 22 个是新的。USH2A 和 MYO7A 分别导致大多数 II 型和 I 型 Usher 综合征病例。BBS1 变异是 Bardet-Biedl 综合征 52.6%家庭的病因。在 99 名患者(198 只眼)的基线和最后一次就诊时获得了最佳矫正视力(BCVA)记录,中位随访时间为 62.0 个月。基线时平均 BCVA 为 56.5 ETDRS 字母(Snellen 等效值约为 20/80),在最后一次可获得的随访时降至 44.9 ETDRS 字母(Snellen 等效值约为 20/125)(p<0.001)。
这是葡萄牙首例多中心研究,描述了综合征性 RP 的遗传特征,从而更好地了解了这一异质性疾病群体。在这个大型葡萄牙队列中,Usher 和 Bardet-Biedl 综合征被发现是最常见的综合征性 RP 类型。获得了高的诊断率,突出了当前基因检测能力在为大多数受影响个体提供分子诊断方面的优势。这对确定疾病相关预后和为葡萄牙综合征性 RP 患者提供有针对性的遗传咨询具有重要意义。