Gouiza Ismail, Hechmi Meriem, Zioudi Abir, Dallali Hamza, Kheriji Nadia, Charif Majida, Le Mao Morgane, Galai Said, Kraoua Lilia, Ben Youssef-Turki Ilhem, Kraoua Ichraf, Lenaers Guy, Kefi Rym
University of Angers, MitoLab Team, Unité MitoVasc, UMR CNRS (Unité mixte de recherche Centre national de la recherche scientifique) 6015 INSERM (Institut national de la santé et de la recherche médicale) U1083, SFR ICAT, University of Angers, Angers, France.
Laboratory of Biomedical Genomics and Oncogenetics, Institut Pasteur de Tunis, Tunis, Tunisia.
Front Genet. 2024 Jan 12;14:1259826. doi: 10.3389/fgene.2023.1259826. eCollection 2023.
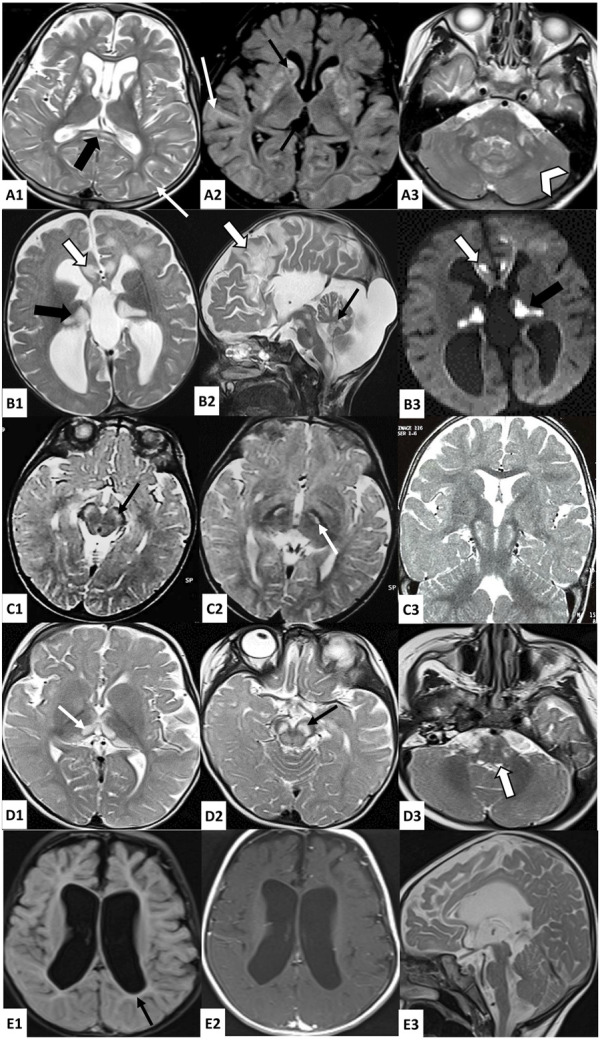
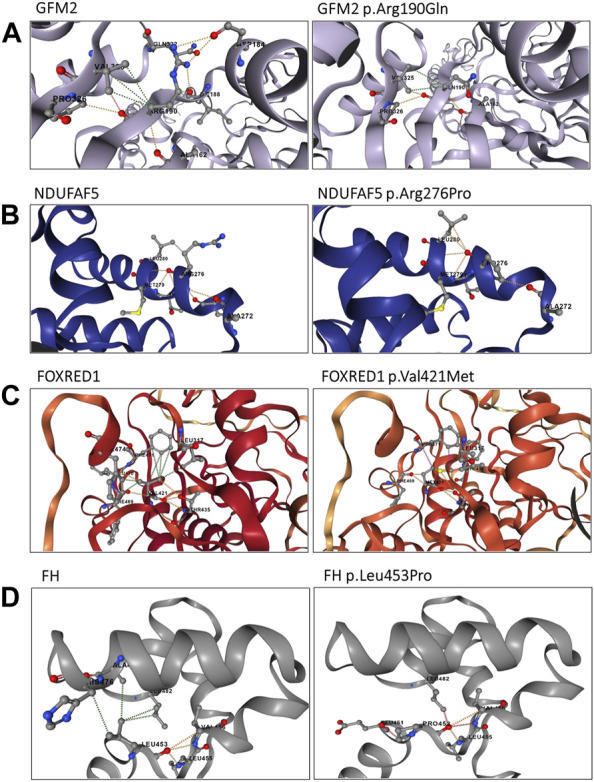
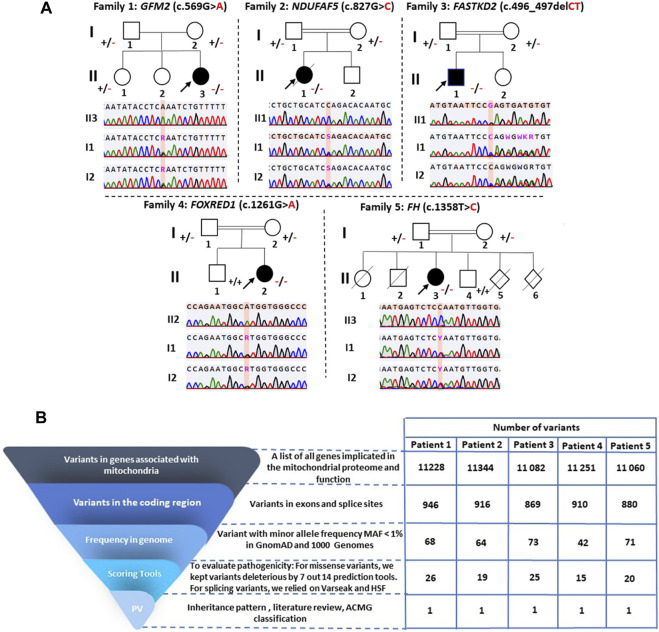
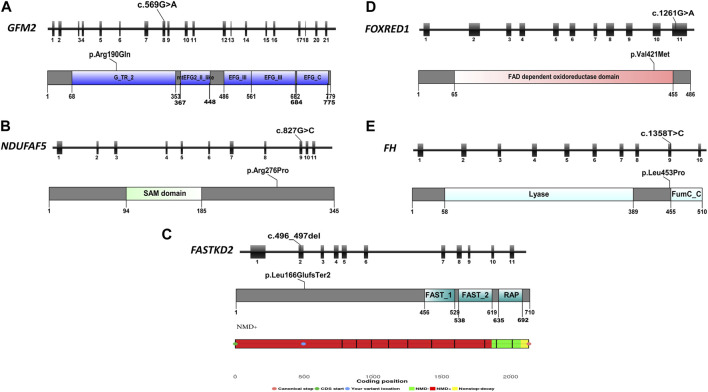
Inherited mitochondrial diseases are the most common group of metabolic disorders caused by a defect in oxidative phosphorylation. They are characterized by a wide clinical and genetic spectrum and can manifest at any age. In this study, we established novel phenotype-genotype correlations between the clinical and molecular features of a cohort of Tunisian patients with mitochondrial diseases. Whole-exome sequencing was performed on five Tunisian patients with suspected mitochondrial diseases. Then, a combination of filtering and bioinformatics prediction tools was utilized to assess the pathogenicity of genetic variations. Sanger sequencing was subsequently performed to confirm the presence of potential deleterious variants in the patients and verify their segregation within families. Structural modeling was conducted to study the effect of novel variants on the protein structure. We identified two novel homozygous variants in (c.827G>C; p.Arg276Pro) and (c.496_497del; p.Leu166GlufsTer2) associated with a severe clinical form of Leigh and Leigh-like syndromes, respectively. Our results further disclosed two variants unreported in North Africa, in (c.569G>A; p.Arg190Gln) and (c.1261G>A; p.Val421Met) genes, and we described the first case of fumaric aciduria in a Tunisian patient harboring the c.1358T>C; p.Leu453Pro variant. Our study expands the mutational and phenotypic spectrum of mitochondrial diseases in Tunisia and highlights the importance of next-generation sequencing to decipher the pathomolecular mechanisms responsible for these disorders in an admixed population.
遗传性线粒体疾病是由氧化磷酸化缺陷引起的最常见的一组代谢紊乱疾病。它们具有广泛的临床和遗传谱,可在任何年龄出现。在本研究中,我们在一组突尼斯线粒体疾病患者的临床和分子特征之间建立了新的表型-基因型相关性。对五名疑似线粒体疾病的突尼斯患者进行了全外显子组测序。然后,利用过滤和生物信息学预测工具的组合来评估基因变异的致病性。随后进行桑格测序以确认患者中潜在有害变异的存在,并验证其在家族中的分离情况。进行了结构建模以研究新变异对蛋白质结构的影响。我们在 (c.827G>C;p.Arg276Pro) 和 (c.496_497del;p.Leu166GlufsTer2) 中分别鉴定出两个与严重临床形式的 Leigh 综合征和 Leigh 样综合征相关的新纯合变异。我们的结果进一步揭示了在北非未报道的两个变异,分别在 (c.569G>A;p.Arg190Gln) 和 (c.1261G>A;p.Val421Met) 基因中,并且我们描述了第一例携带 c.1358T>C;p.Leu453Pro 变异的突尼斯患者患富马酸尿症的病例。我们的研究扩展了突尼斯线粒体疾病的突变和表型谱,并强调了下一代测序在解读混合人群中这些疾病致病分子机制方面的重要性。