Astner-Rohracher Alexandra, Mauritz Matthias, Leitinger Markus, Rossini Fabio, Kalss Gudrun, Neuray Caroline, Retter Elisabeth, Wortmann Saskia B, Achleitner Melanie T, Mayr Johannes A, Trinka Eugen
Department of Neurology, Neurocritical Care, and Neurorehabilitation, Christian Doppler University Hospital, Centre for Cognitive Neuroscience, Paracelsus Medical University, Salzburg, Austria.
Neuroscience Institute, Christian Doppler University Hospital, Centre for Cognitive Neuroscience Paracelsus Medical University Hospital, Salzburg, Austria.
Front Neurol. 2023 Jan 11;13:1063733. doi: 10.3389/fneur.2022.1063733. eCollection 2022.
New-onset refractory status epilepticus (NORSE) is associated with high morbidity and mortality. Despite extensive work-up, the underlying etiology remains unknown in 50% of affected individuals. Mitochondrial disorders represent rare causes of NORSE. Biallelic variants in were reported as a cause of infantile encephalomyopathy with refractory epilepsy.
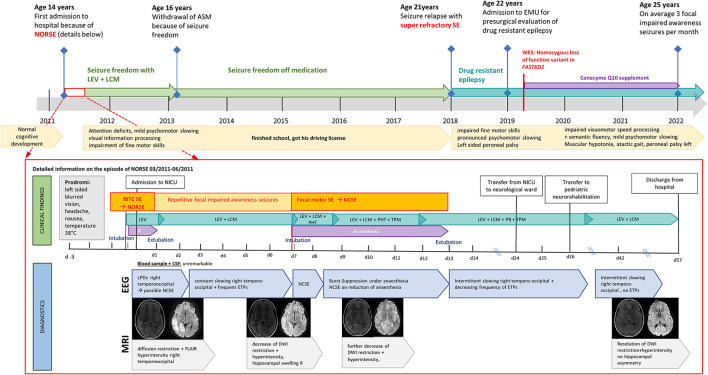
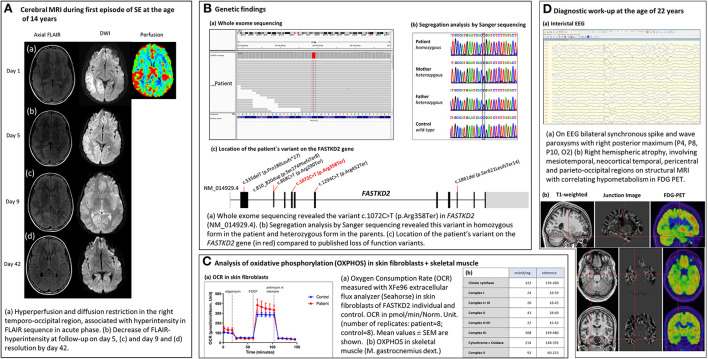
In the study, we report a previously healthy 14-year-old with a new, homozygous variant presenting with NORSE. Following a seizure-free period of 7 years, he experienced another super-refractory SE and subsequently developed drug-resistant focal epilepsy, mild myopathy, optic atrophy, and discrete psychomotor slowing. Structural MRI at the time of NORSE showed right temporo-parieto-occipital FLAIR hyperintensity and diffusion restriction, with extensive right hemispheric atrophy at the age of 22 years. Whole-exome sequencing revealed a novel homozygous loss of function variant [c.(1072C>T);(1072C>T)] [p.(Arg358Ter);(Arg358Ter)] in (NM_001136193), resulting in a premature termination codon in the protein-coding region and loss of function of FASTKD2. Oxidative phosphorylation (OXPHOS) in muscle and skin fibroblasts was unremarkable.
This is the first case of a normally developed adolescent with a new homozygous loss of function variant in , manifesting with NORSE. The phenotypical spectrum of FASTKD2-related mitochondrial disease is heterogeneous, ranging from recurrent status epilepticus and refractory focal epilepsy in an adolescent with normal cognitive development to severe forms of infantile mitochondrial encephalopathy. Although mitochondrial diseases are rare causes of NORSE, clinical features such as young age at onset and multi-system involvement should trigger genetic testing. Early diagnosis is essential for counseling and treatment considerations.
新发难治性癫痫持续状态(NORSE)与高发病率和死亡率相关。尽管进行了广泛检查,但50%的患者潜在病因仍不明。线粒体疾病是NORSE的罕见病因。据报道, 中的双等位基因变异是婴儿期难治性癫痫性脑病的病因。
在本研究中,我们报告了一名既往健康的14岁青少年,其出现新的纯合 变异并伴有NORSE。在7年无癫痫发作期后,他再次出现超难治性癫痫持续状态,随后发展为耐药性局灶性癫痫、轻度肌病、视神经萎缩和明显的精神运动迟缓。NORSE发作时的结构MRI显示右侧颞顶枕叶液体衰减反转恢复序列(FLAIR)高信号和弥散受限,22岁时右侧大脑半球广泛萎缩。全外显子测序揭示了 (NM_001136193)中一个新的纯合功能缺失变异[c.(1072C>T);(1072C>T)][p.(Arg358Ter);(Arg358Ter)],导致蛋白质编码区出现过早终止密码子,FASTKD2功能丧失。肌肉和皮肤成纤维细胞中的氧化磷酸化(OXPHOS)无异常。
这是首例正常发育的青少年因 中出现新的纯合功能缺失变异而表现为NORSE的病例。FASTKD2相关线粒体疾病的表型谱具有异质性,从认知发育正常的青少年复发性癫痫持续状态和难治性局灶性癫痫到严重形式的婴儿线粒体脑病。尽管线粒体疾病是NORSE的罕见病因,但起病年龄小和多系统受累等临床特征应引发基因检测。早期诊断对于咨询和治疗考量至关重要。