Center of Medical Genetics, Faculty of Medicine and Health Sciences, University of Antwerp and Antwerp University Hospital, Antwerp, Belgium.
Department of Clinical Genetics, Radboud University Medical Center, Nijmegen, Netherlands.
J Med Genet. 2024 Mar 21;61(4):363-368. doi: 10.1136/jmg-2023-109151.
encodes an intracellular inhibitor of the bone morphogenetic protein (BMP) signalling pathway. Until now, rare heterozygous loss-of-function variants in were demonstrated to increase the risk of disparate clinical disorders including cardiovascular disease, craniosynostosis and radioulnar synostosis. Only two unrelated patients harbouring biallelic variants presenting a complex cardiovascular phenotype and facial dysmorphism have been described.
Here, we present the first two patients with craniosynostosis harbouring homozygous variants. The male probands, both born to healthy consanguineous parents, were diagnosed with metopic synostosis and bilateral or unilateral radioulnar synostosis. Additionally, one proband had global developmental delay. Echocardiographic evaluation did not reveal cardiac or outflow tract abnormalities.
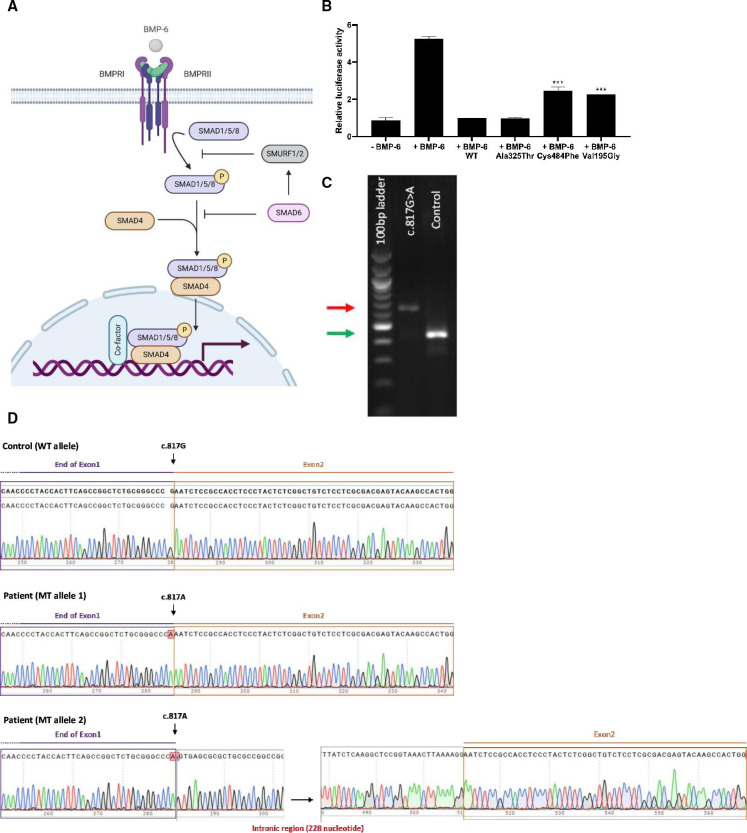
The novel missense (c.[584T>G];[584T>G], p.[(Val195Gly)];[(Val195Gly)]) and missense/splice-site variant (c.[817G>A];[817G>A], r.[(817g>a,817delins[a;817+2_817+228])];[(817g>a,817delins[a;817+2_817+228])], p.[(Glu273Lys,Glu273Serfs72)];[(Glu273Lys,Glu273Serfs72)]) both locate in the functional MH1 domain of the protein and have not been reported in gnomAD database. Functional analyses of the variants showed reduced inhibition of BMP signalling or abnormal splicing, respectively, consistent with a hypomorphic mechanism of action.
Our data expand the spectrum of variants and phenotypic spectrum associated with homozygous variants of to include craniosynostosis.
编码骨形态发生蛋白(BMP)信号通路的细胞内抑制剂。到目前为止,已经证明 在 中罕见的杂合性功能丧失变体增加了包括心血管疾病、颅缝早闭和桡尺骨融合在内的不同临床疾病的风险。仅描述了两名携带表现出复杂心血管表型和面部畸形的 双等位基因 变体的无关患者。
在这里,我们介绍了两名患有颅缝早闭的患者,他们均携带纯合子 变体。男性先证者均出生于健康的近亲父母,被诊断为额缝早闭和双侧或单侧桡尺骨融合。此外,一名先证者有全面发育迟缓。超声心动图评估未发现心脏或流出道异常。
新的错义突变(c.[584T>G];[584T>G],p.[(Val195Gly)];[(Val195Gly)])和错义/剪接位点变体(c.[817G>A];[817G>A],r.[(817g>a,817delins[a;817+2_817+228])];[(817g>a,817delins[a;817+2_817+228])],p.[(Glu273Lys,Glu273Serfs72)];[(Glu273Lys,Glu273Serfs72)])均位于蛋白质的功能 MH1 结构域中,在 gnomAD 数据库中未报道过。变体的功能分析分别显示 BMP 信号抑制减少或异常剪接,与功能减退的作用机制一致。
我们的数据扩展了与 纯合变体相关的变体和表型谱,包括颅缝早闭。