Urzhumtsev Alexandre, Adams Paul, Afonine Pavel
Centre for Integrative Biology, Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS-INSERM-UdS, 1 rue Laurent Fries, BP 10142, 67404 Illkirch, France.
Molecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, California, USA.
Acta Crystallogr A Found Adv. 2024 Mar 1;80(Pt 2):194-201. doi: 10.1107/S2053273324000299. Epub 2024 Feb 9.
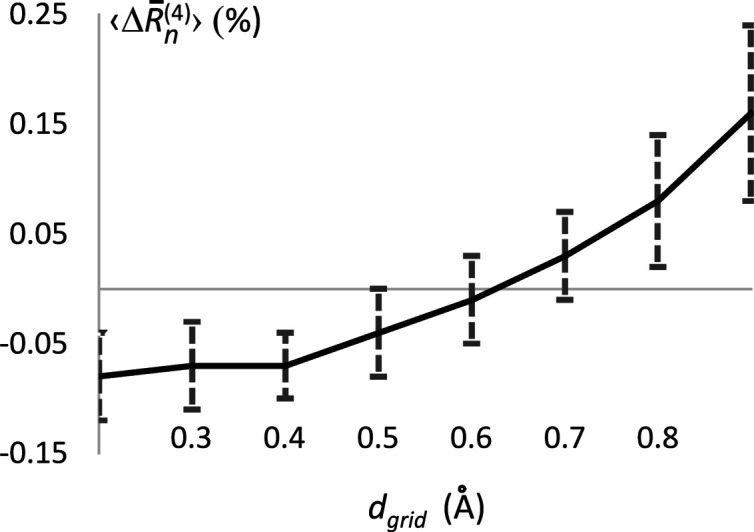
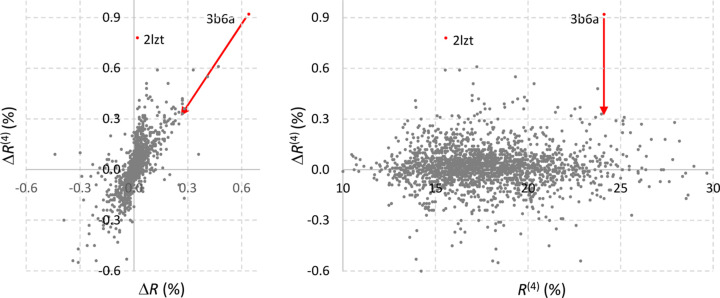
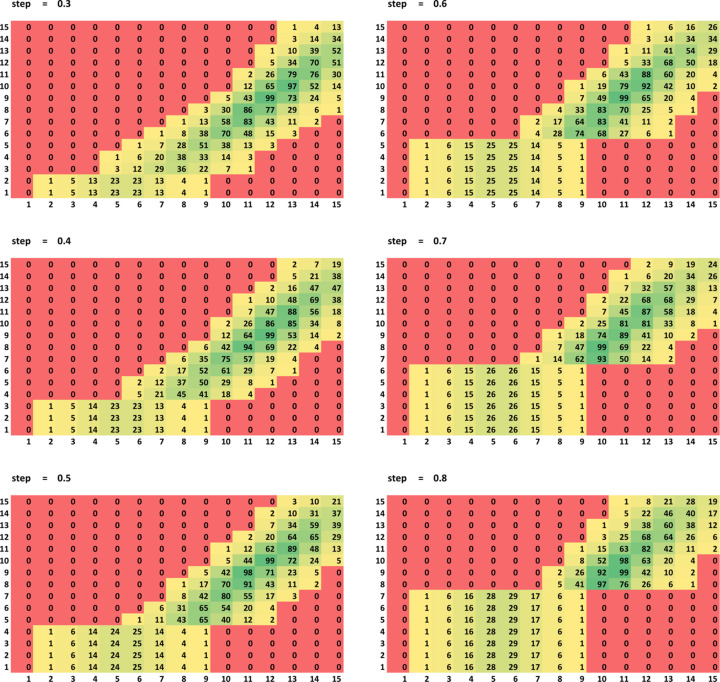
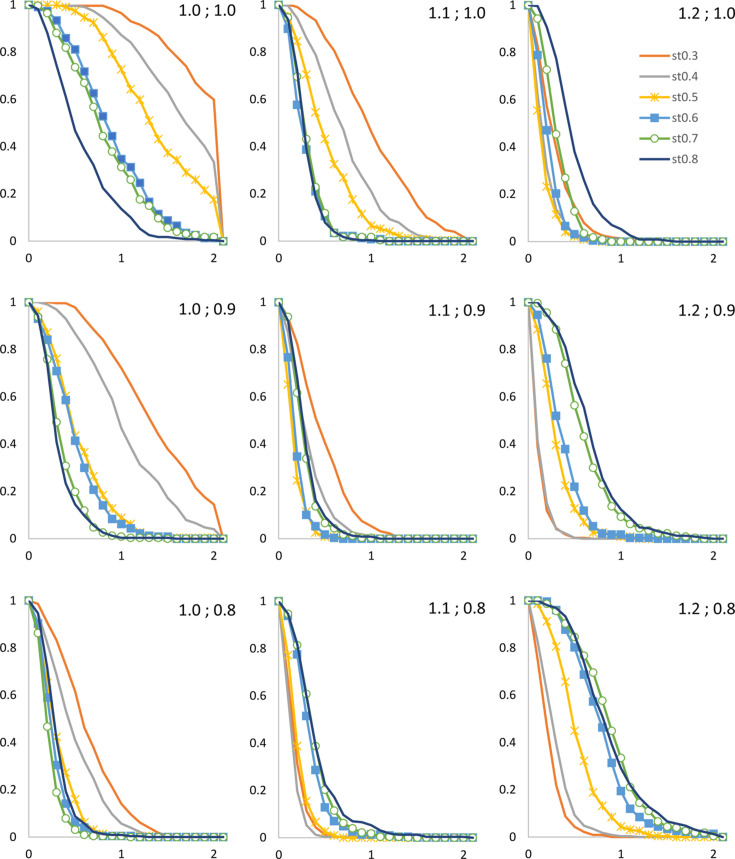
The bulk solvent is a major component of biomacromolecular crystals that contributes significantly to the observed diffraction intensities. Accurate modelling of the bulk solvent has been recognized as important for many crystallographic calculations. Owing to its simplicity and modelling power, the flat (mask-based) bulk-solvent model is used by most modern crystallographic software packages to account for disordered solvent. In this model, the bulk-solvent contribution is defined by a binary mask and a scale (scattering) function. The mask is calculated on a regular grid using the atomic model coordinates and their chemical types. The grid step and two radii, solvent and shrinkage, are the three parameters that govern the mask calculation. They are highly correlated and their choice is a compromise between the computer time needed to calculate the mask and the accuracy of the mask. It is demonstrated here that this choice can be optimized using a unique value of 0.6 Å for the grid step irrespective of the data resolution, and the radii values adjusted correspondingly. The improved values were tested on a large sample of Protein Data Bank entries derived from X-ray diffraction data and are now used in the computational crystallography toolbox (CCTBX) and in Phenix as the default choice.
大量溶剂是生物大分子晶体的主要成分,对观察到的衍射强度有显著贡献。准确模拟大量溶剂已被认为对许多晶体学计算很重要。由于其简单性和建模能力,大多数现代晶体学软件包都使用平面(基于掩模)大量溶剂模型来处理无序溶剂。在该模型中,大量溶剂的贡献由二元掩模和标度(散射)函数定义。掩模是使用原子模型坐标及其化学类型在规则网格上计算的。网格步长以及溶剂半径和收缩半径这两个半径是控制掩模计算的三个参数。它们高度相关,其选择是在计算掩模所需的计算机时间和掩模的准确性之间进行折衷。本文证明,无论数据分辨率如何,使用0.6 Å的唯一网格步长值并相应调整半径值,可以优化这种选择。在从X射线衍射数据得出的大量蛋白质数据库条目中对改进后的值进行了测试,现在它们在计算晶体学工具箱(CCTBX)和Phenix中作为默认选择使用。