Department of Microbial Ecology, The Netherlands Institute of Ecology (NIOO-KNAW), P.O. Box 50, 6700 AB Wageningen, The Netherlands.
Viruses. 2024 Feb 8;16(2):270. doi: 10.3390/v16020270.
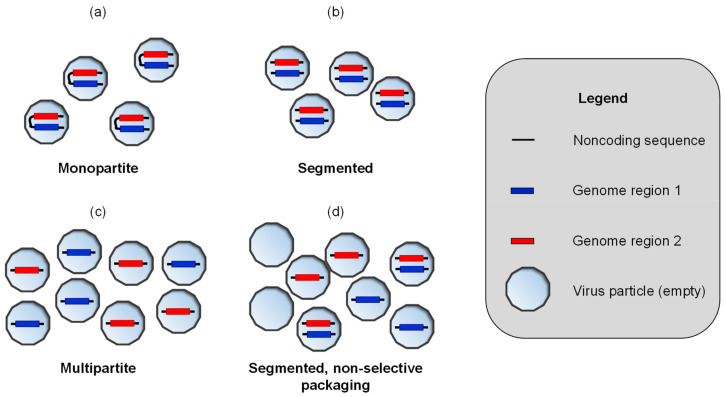
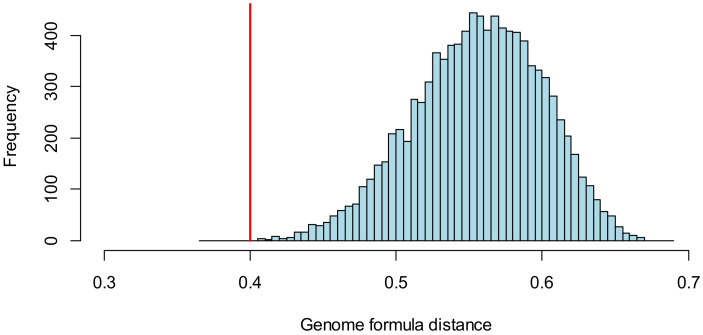
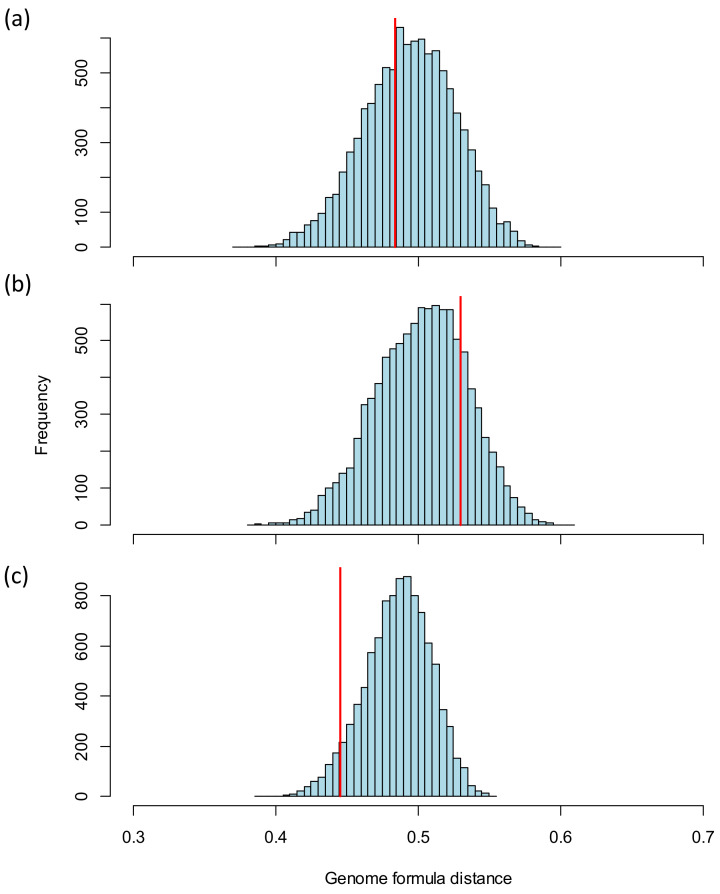
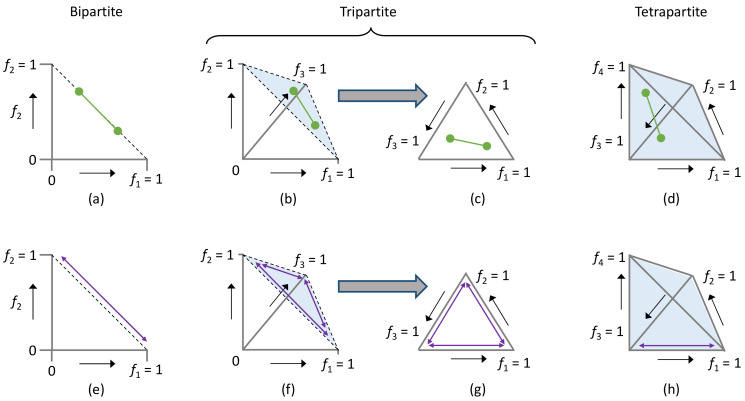
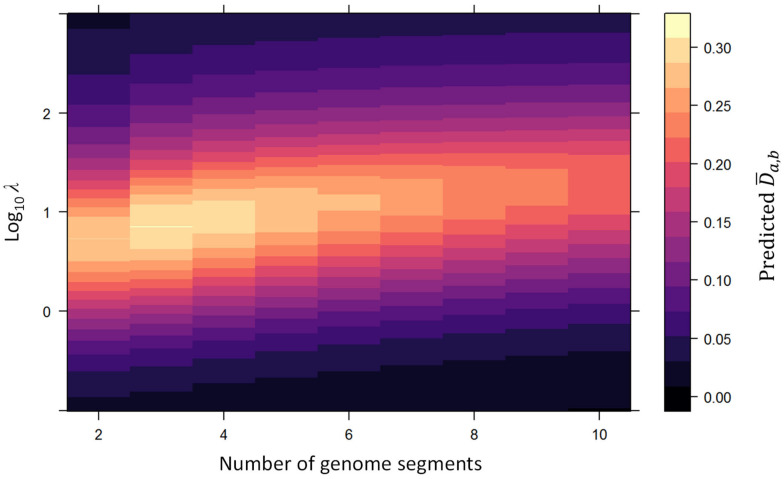
When viruses have segmented genomes, the set of frequencies describing the abundance of segments is called the genome formula. The genome formula is often unbalanced and highly variable for both segmented and multipartite viruses. A growing number of studies are quantifying the genome formula to measure its effects on infection and to consider its ecological and evolutionary implications. Different approaches have been reported for analyzing genome formula data, including qualitative description, applying standard statistical tests such as ANOVA, and customized analyses. However, these approaches have different shortcomings, and test assumptions are often unmet, potentially leading to erroneous conclusions. Here, we address these challenges, leading to a threefold contribution. First, we propose a simple metric for analyzing genome formula variation: the genome formula distance. We describe the properties of this metric and provide a framework for understanding metric values. Second, we explain how this metric can be applied for different purposes, including testing for genome-formula differences and comparing observations to a reference genome formula value. Third, we re-analyze published data to illustrate the applications and weigh the evidence for previous conclusions. Our re-analysis of published datasets confirms many previous results but also provides evidence that the genome formula can be carried over from the inoculum to the virus population in a host. The simple procedures we propose contribute to the robust and accessible analysis of genome-formula data.
当病毒具有分段基因组时,描述片段丰度的频率集被称为基因组公式。对于分段和多部分病毒,基因组公式通常是不平衡且高度可变的。越来越多的研究正在量化基因组公式,以衡量其对感染的影响,并考虑其生态和进化意义。已经报道了多种分析基因组公式数据的方法,包括定性描述、应用方差分析 (ANOVA) 等标准统计检验以及定制分析。然而,这些方法各有缺点,并且检验假设通常得不到满足,可能导致错误的结论。在这里,我们解决了这些挑战,做出了三方面的贡献。首先,我们提出了一种用于分析基因组公式变异的简单度量标准:基因组公式距离。我们描述了这个度量标准的性质,并提供了一个理解度量值的框架。其次,我们解释了如何将该度量标准应用于不同的目的,包括测试基因组公式的差异和将观察结果与参考基因组公式值进行比较。第三,我们重新分析了已发表的数据,以说明这些应用,并权衡对先前结论的证据。我们对已发表数据集的重新分析证实了许多先前的结果,但也提供了证据表明,基因组公式可以从接种物传递到宿主中的病毒群体。我们提出的简单程序有助于对基因组公式数据进行稳健且易于访问的分析。