State Key Laboratory of Tree Genetics and Breeding, National Engineering Research Center of Tree Breeding and Ecological Restoration, Beijing Advanced Innovation Center for Tree Breeding by Molecular Design, National Engineering Laboratory for Tree Breeding, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants, Ministry of Education, College of Biological Sciences and Technology, Beijing Forestry University, Beijing 100083, China.
Key Laboratory of Crop Genetic Improvement & Ecology and Physiology, Institute of Crop Germplasm Resources, Shandong Academy of Agricultural Sciences, Ji'nan 250100, China.
Plant Physiol. 2024 Apr 30;195(1):652-670. doi: 10.1093/plphys/kiae078.
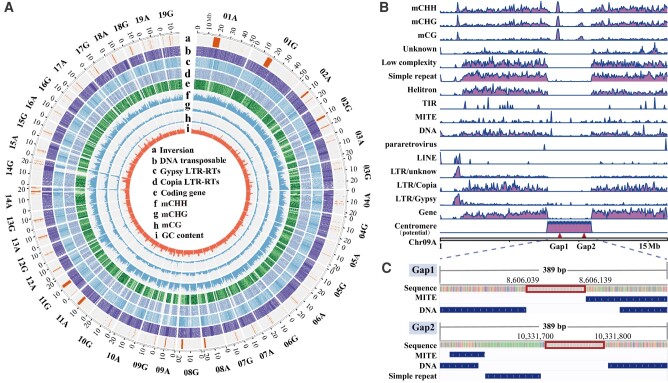
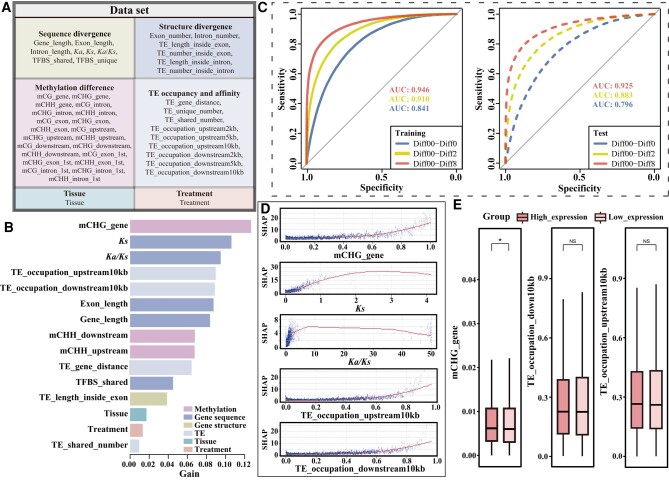
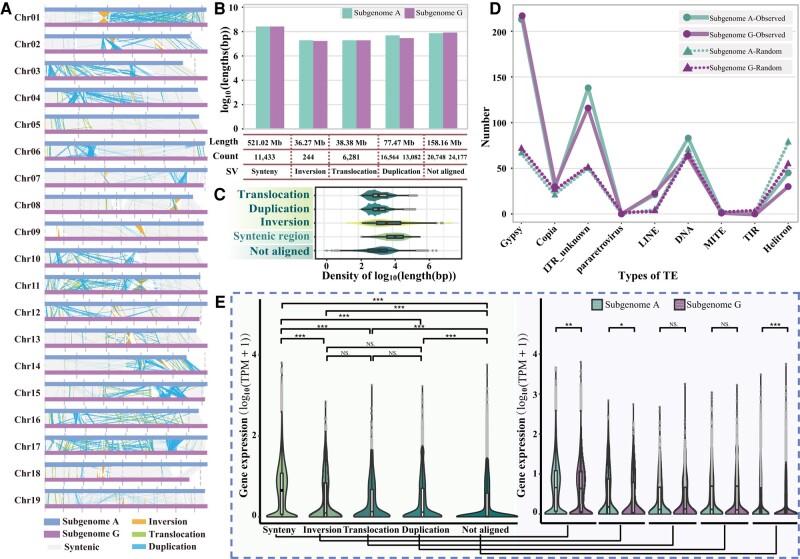
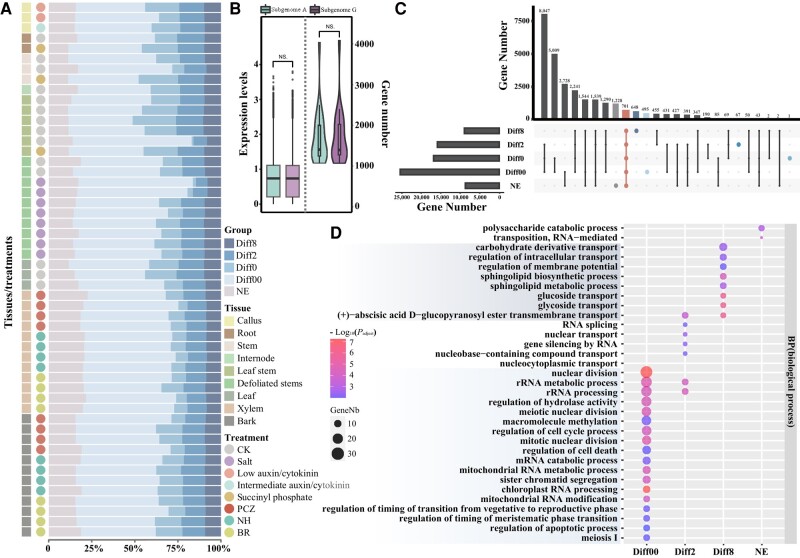
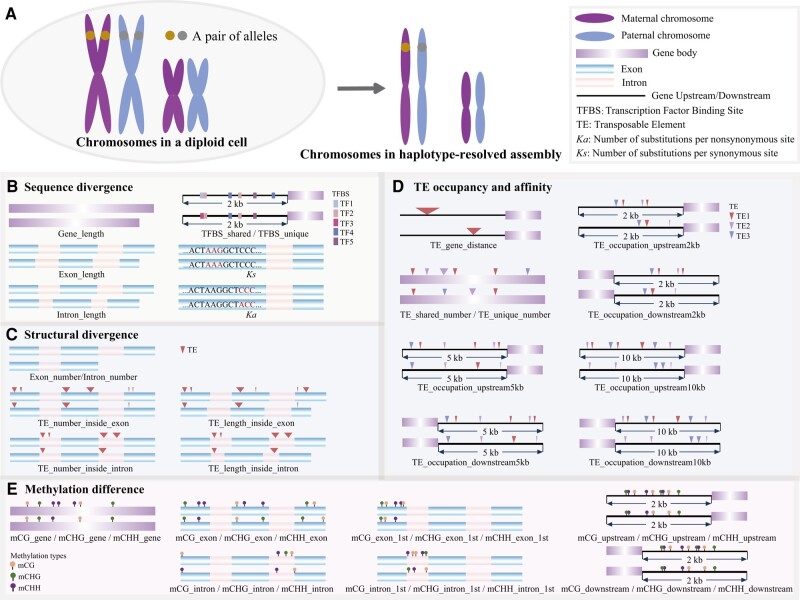
Poplar (Populus) is a well-established model system for tree genomics and molecular breeding, and hybrid poplar is widely used in forest plantations. However, distinguishing its diploid homologous chromosomes is difficult, complicating advanced functional studies on specific alleles. In this study, we applied a trio-binning design and PacBio high-fidelity long-read sequencing to obtain haplotype-phased telomere-to-telomere genome assemblies for the 2 parents of the well-studied F1 hybrid "84K" (Populus alba × Populus tremula var. glandulosa). Almost all chromosomes, including the telomeres and centromeres, were completely assembled for each haplotype subgenome apart from 2 small gaps on one chromosome. By incorporating information from these haplotype assemblies and extensive RNA-seq data, we analyzed gene expression patterns between the 2 subgenomes and alleles. Transcription bias at the subgenome level was not uncovered, but extensive-expression differences were detected between alleles. We developed machine-learning (ML) models to predict allele-specific expression (ASE) with high accuracy and identified underlying genome features most highly influencing ASE. One of our models with 15 predictor variables achieved 77% accuracy on the training set and 74% accuracy on the testing set. ML models identified gene body CHG methylation, sequence divergence, and transposon occupancy both upstream and downstream of alleles as important factors for ASE. Our haplotype-phased genome assemblies and ML strategy highlight an avenue for functional studies in Populus and provide additional tools for studying ASE and heterosis in hybrids.
杨属(Populus)是树木基因组学和分子育种的成熟模式系统,杂交杨广泛用于人工林种植。然而,区分其二倍体同源染色体较为困难,这使得对特定等位基因的高级功能研究变得复杂。在这项研究中,我们应用了三亲 binning 设计和 PacBio 高保真长读测序,为经过深入研究的 F1 杂种“84K”(银白杨×毛果杨)的 2 个亲本获得了单体型相位端粒到端粒全基因组组装。除了一条染色体上的 2 个小缺口外,每个单体型亚基因组的几乎所有染色体,包括端粒和着丝粒,都被完全组装。通过整合这些单体型组装和广泛的 RNA-seq 数据,我们分析了 2 个亚基因组和等位基因之间的基因表达模式。在亚基因组水平上未发现转录偏向,但在等位基因之间检测到广泛的表达差异。我们开发了机器学习(ML)模型,以高精度预测等位基因特异性表达(ASE),并确定了最能影响 ASE 的潜在基因组特征。我们的模型中有 15 个预测变量,在训练集上的准确率为 77%,在测试集上的准确率为 74%。ML 模型确定了等位基因上下游的基因体 CHG 甲基化、序列分化和转座子占据作为 ASE 的重要因素。我们的单体型相位基因组组装和 ML 策略为杨属的功能研究提供了一个途径,并为研究 ASE 和杂种优势提供了额外的工具。