National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, 41008, Hunan Province, People's Republic of China.
Department of Clinical Laboratory, Xiangya Hospital, Central South University, Changsha, Hunan Province, People's Republic of China.
BMC Genomics. 2024 Feb 27;25(1):216. doi: 10.1186/s12864-024-10146-z.
Shewanella xiamenensis, widely distributed in natural environments, has long been considered as opportunistic pathogen. Recently, significant changes in the resistance spectrum have been observed in S. xiamenensis, due to acquired antibiotic resistance genes. Therefore, a pan-genome analysis was conducted to illuminate the genomic changes in S. xiamenensis.
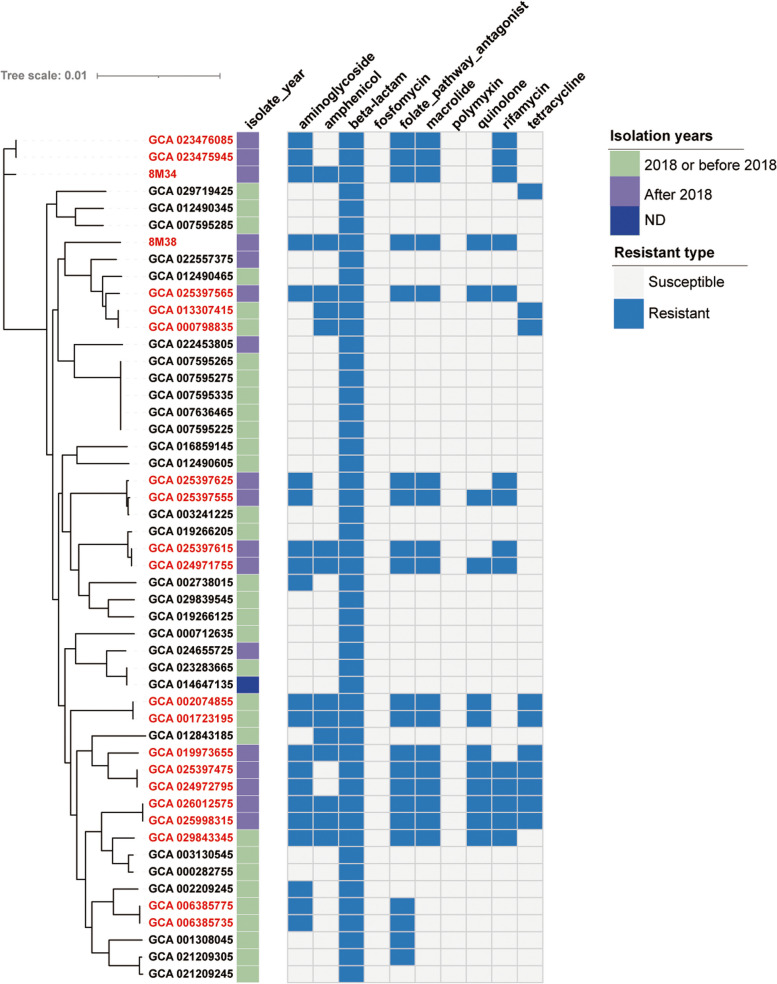
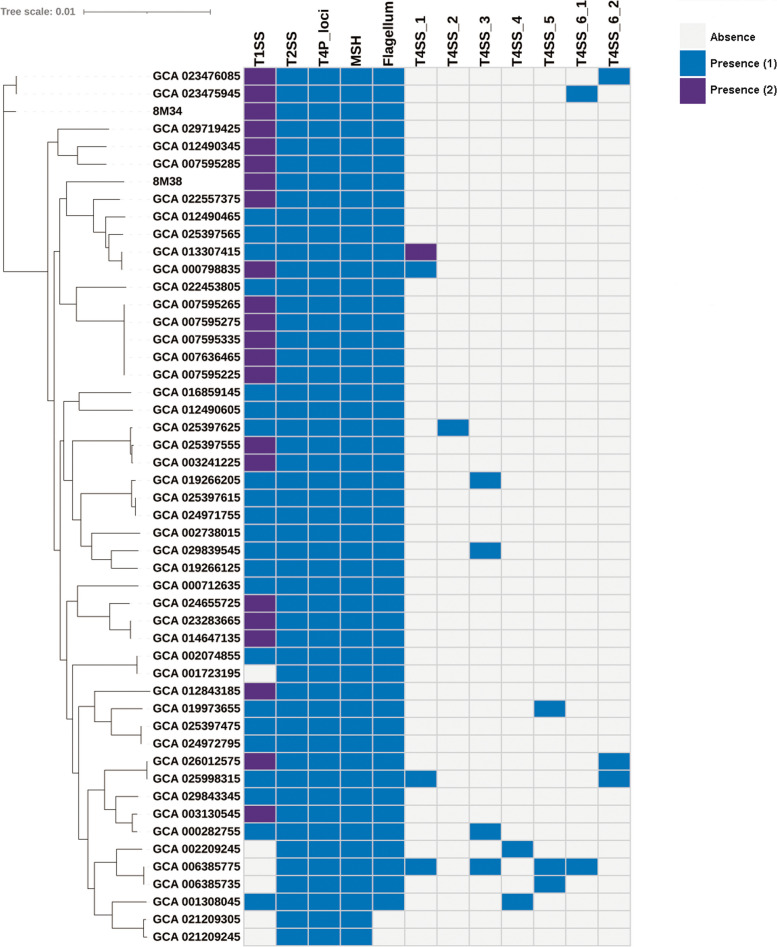
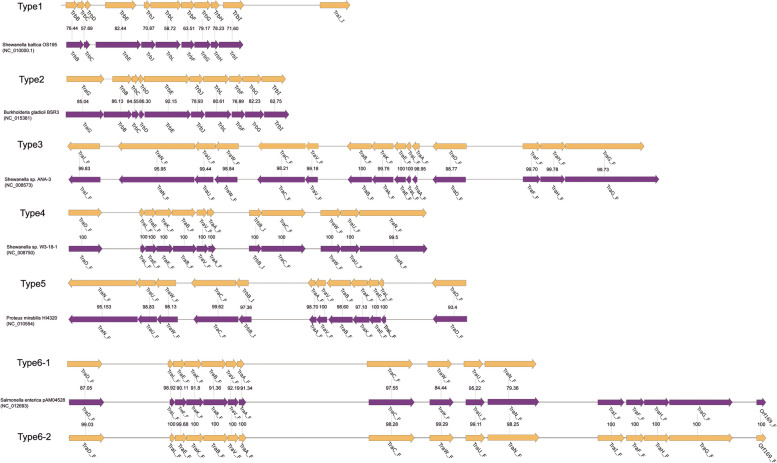
Phylogenetic analysis revealed three major clusters and three singletons, among which close relationship between several strains was discovered, regardless of their host and niches. The "open" genomes with diversity of accessory and strain-specific genomes took advantage towards diversity environments. The purifying selection pressure was the main force on genome evolution, especially in conservative genes. Only 53 gene families were under positive selection pressure. Phenotypic resistance analysis revealed 21 strains were classified as multi-drug resistance (MDR). Ten types of antibiotic resistance genes and two heavy metal resistance operons were discovered in S. xiamenensis. Mobile genetic elements and horizontal gene transfer increased genome diversity and were closely related to MDR strains. S. xiamenensis carried a variety of virulence genes and macromolecular secretion systems, indicating their important roles in pathogenicity and adaptability. Type IV secretion system was discovered in 15 genomes with various sequence structures, indicating it was originated from different donors through horizontal gene transfer.
This study provided with a detailed insight into the changes in the pan-genome of S. xiamenensis, highlighting its capability to acquire new mobile genetic elements and resistance genes for its adaptation to environment and pathogenicity to human and animals.
希瓦氏菌属广泛分布于自然环境中,长期以来被认为是机会致病菌。最近,由于获得了抗生素耐药基因,希瓦氏菌的耐药谱发生了显著变化。因此,进行了全基因组分析以阐明希瓦氏菌的基因组变化。
系统发育分析显示存在三个主要聚类和三个单倍型,其中发现了几个菌株之间的密切关系,无论其宿主和生态位如何。具有多样性的“开放”基因组和菌株特异性基因组有利于多样性的环境。净化选择压力是基因组进化的主要力量,尤其是在保守基因中。只有 53 个基因家族受到正选择压力的影响。表型耐药分析显示 21 株被归类为多药耐药(MDR)。在希瓦氏菌中发现了 10 种抗生素耐药基因和 2 个重金属耐药操纵子。移动遗传元件和水平基因转移增加了基因组的多样性,并与 MDR 菌株密切相关。希瓦氏菌携带多种毒力基因和大分子分泌系统,表明它们在致病性和适应性方面的重要作用。在 15 个基因组中发现了多种序列结构的 IV 型分泌系统,表明它是通过水平基因转移从不同供体起源的。
本研究深入了解了希瓦氏菌全基因组的变化,强调了其获取新的移动遗传元件和耐药基因的能力,以适应环境和对人类和动物的致病性。