Department of Pharmaceutical Chemistry, Amrita School of Pharmacy, Amrita Vishwa Vidyapeetham, AIMS Health Sciences Campus, Kochi, India.
Department of Pharmaceutical Chemistry, School of Pharmaceutical Education and Research, Jamia Hamdard, New Delhi, India.
Sci Rep. 2024 Feb 28;14(1):4868. doi: 10.1038/s41598-024-55628-y.
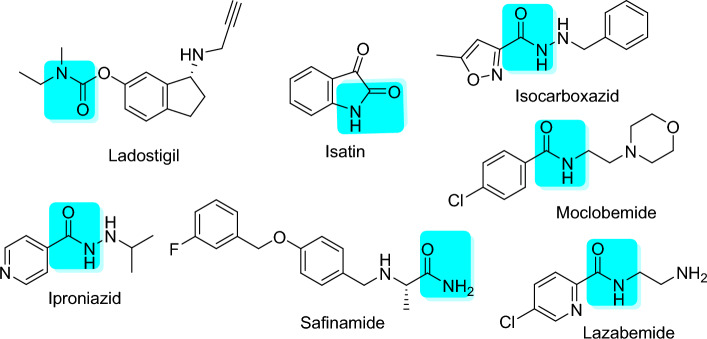
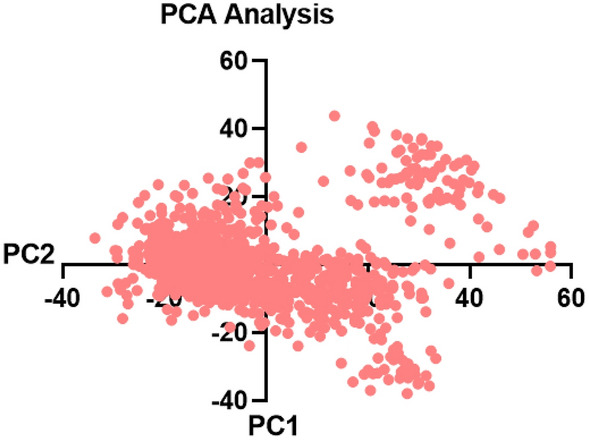
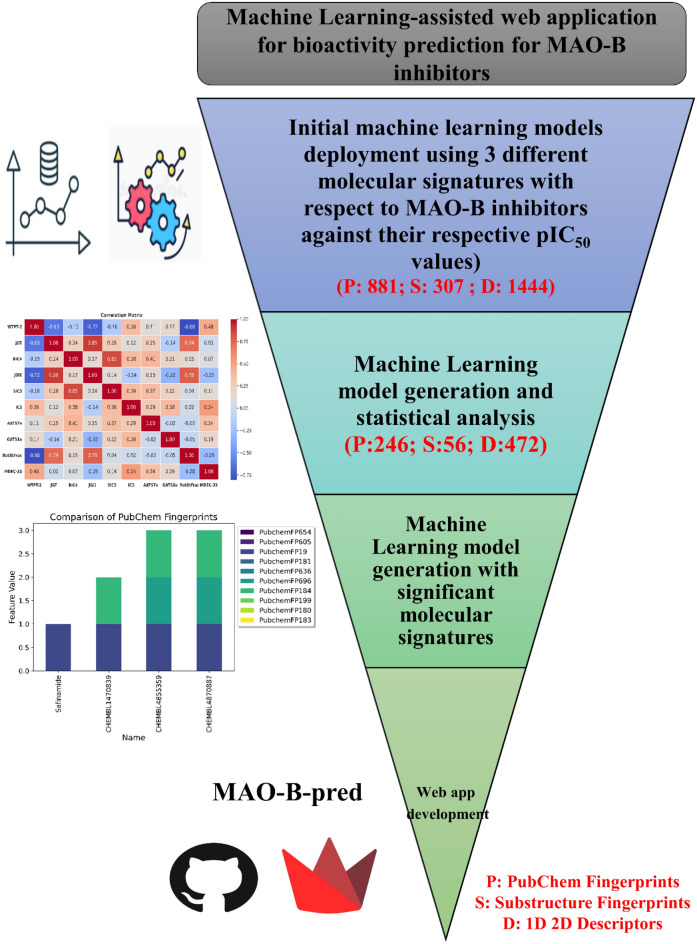
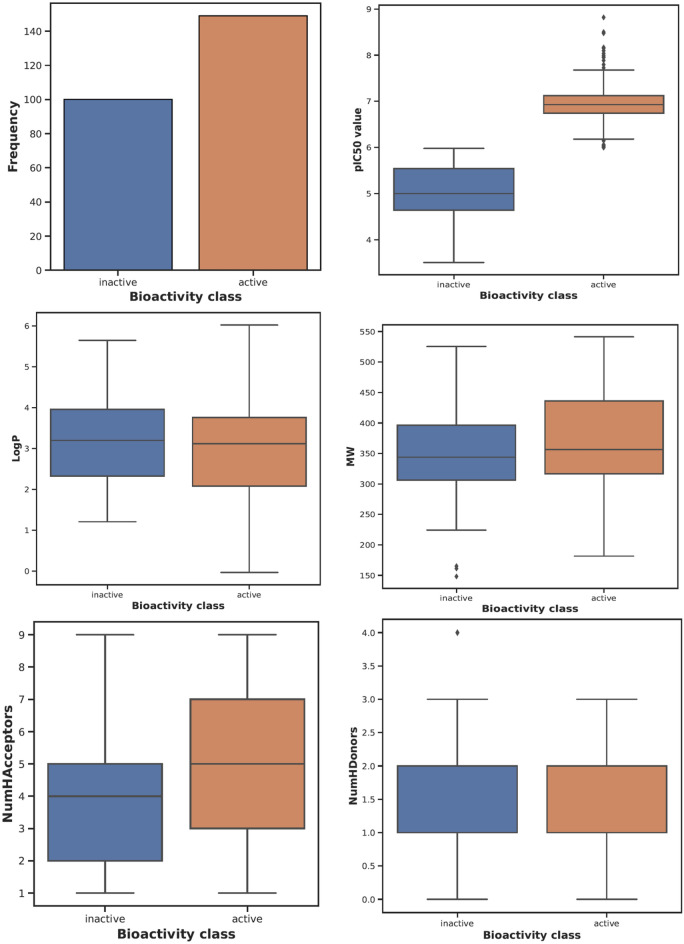
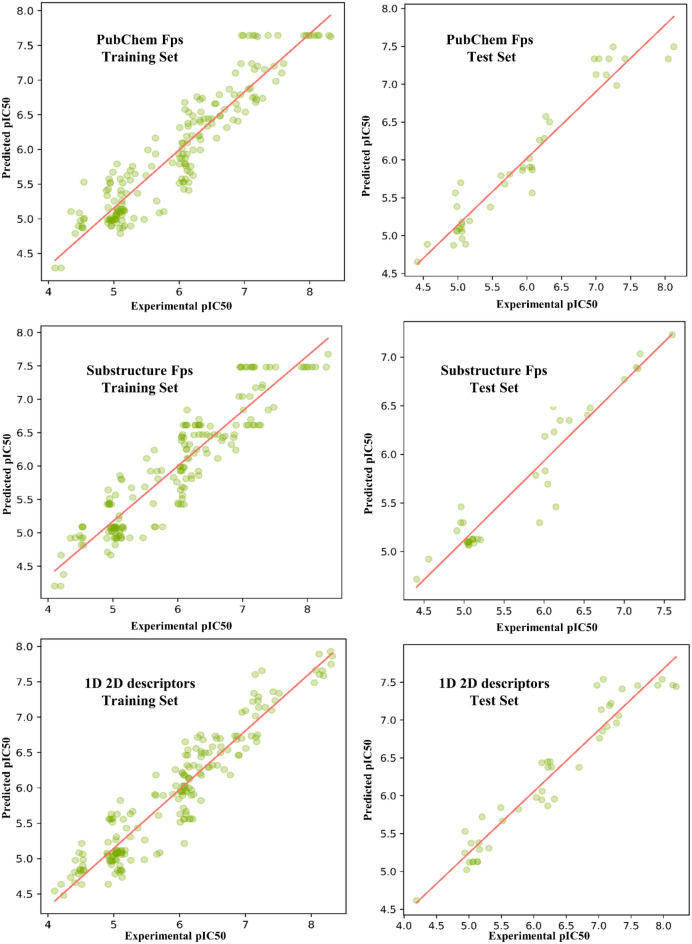
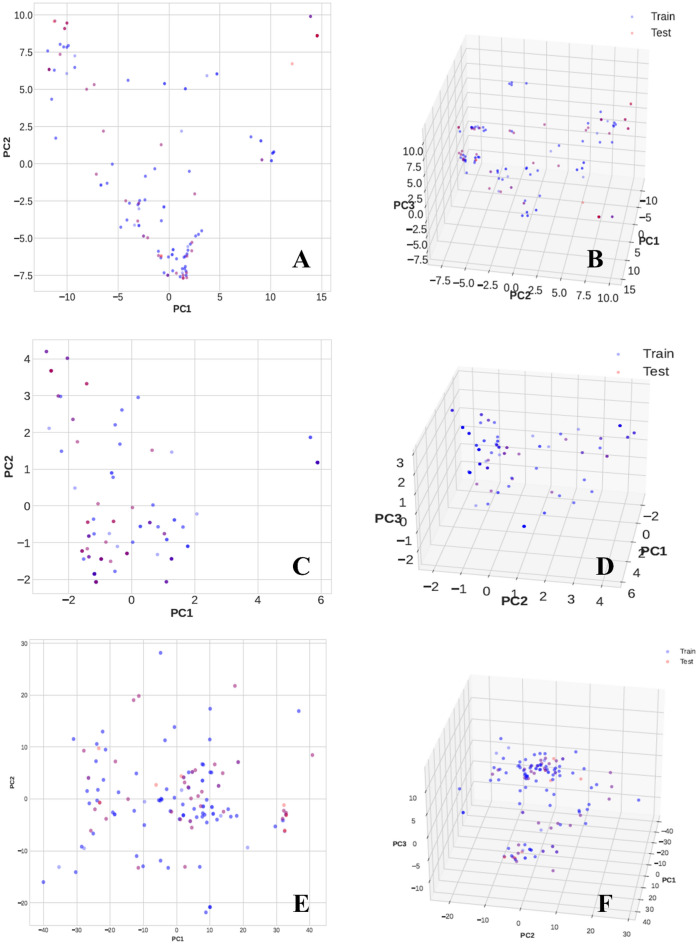
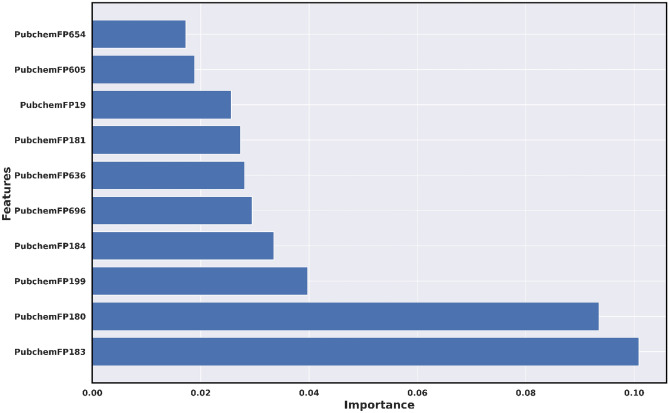
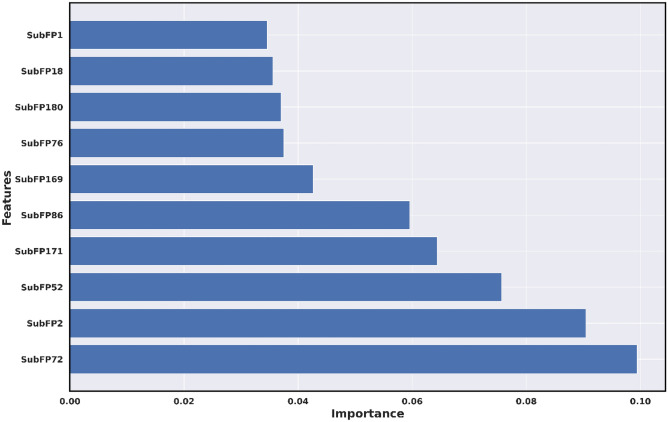
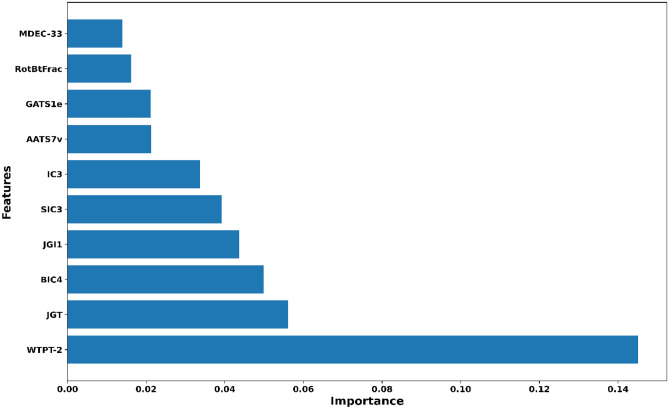
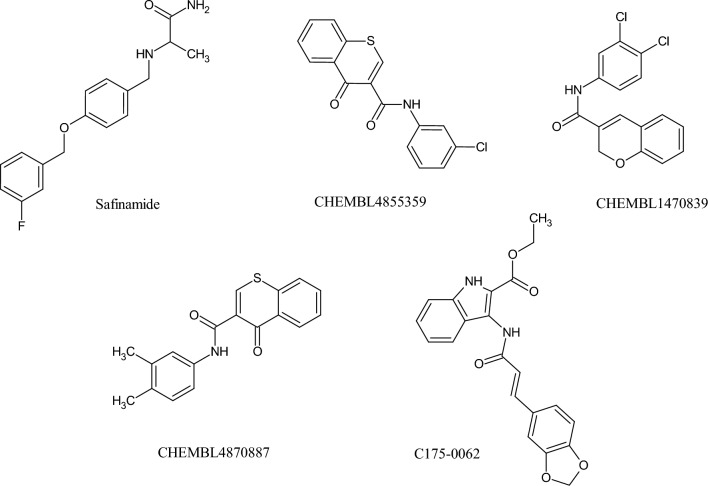
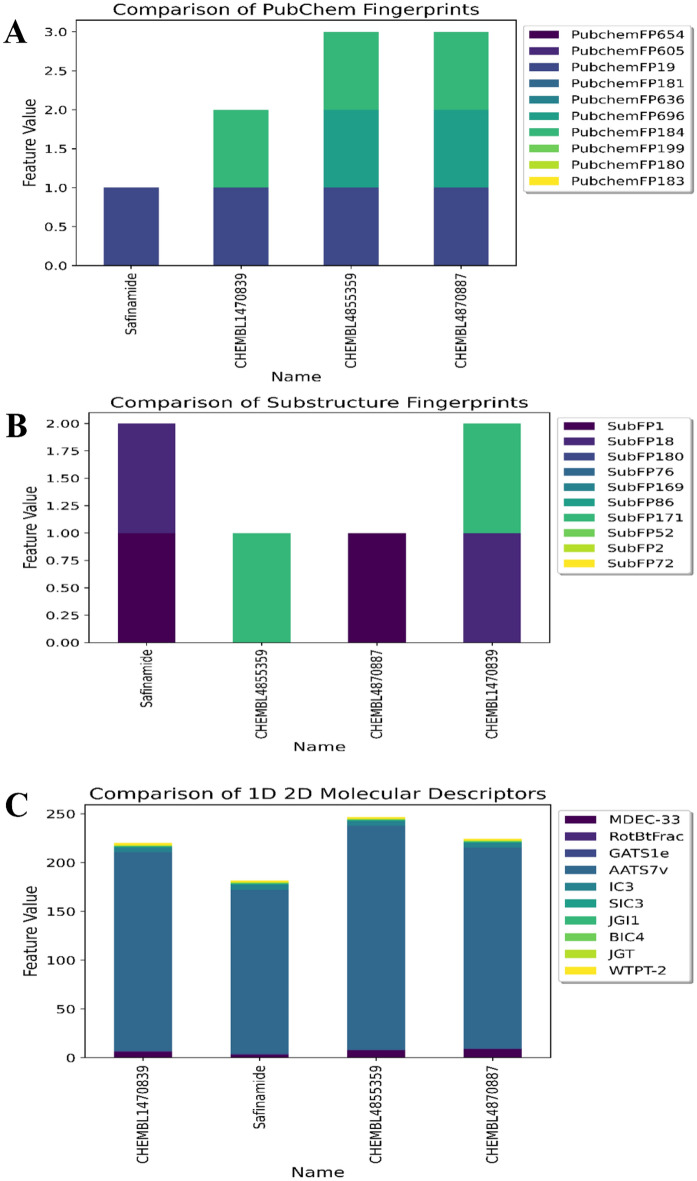
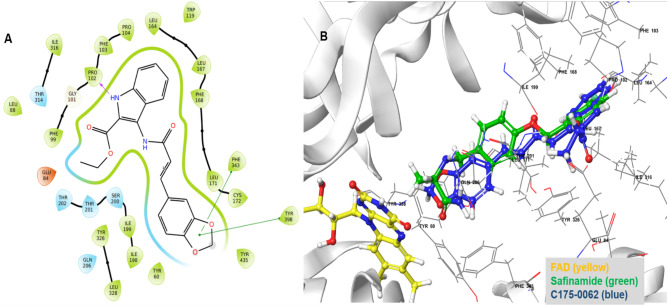
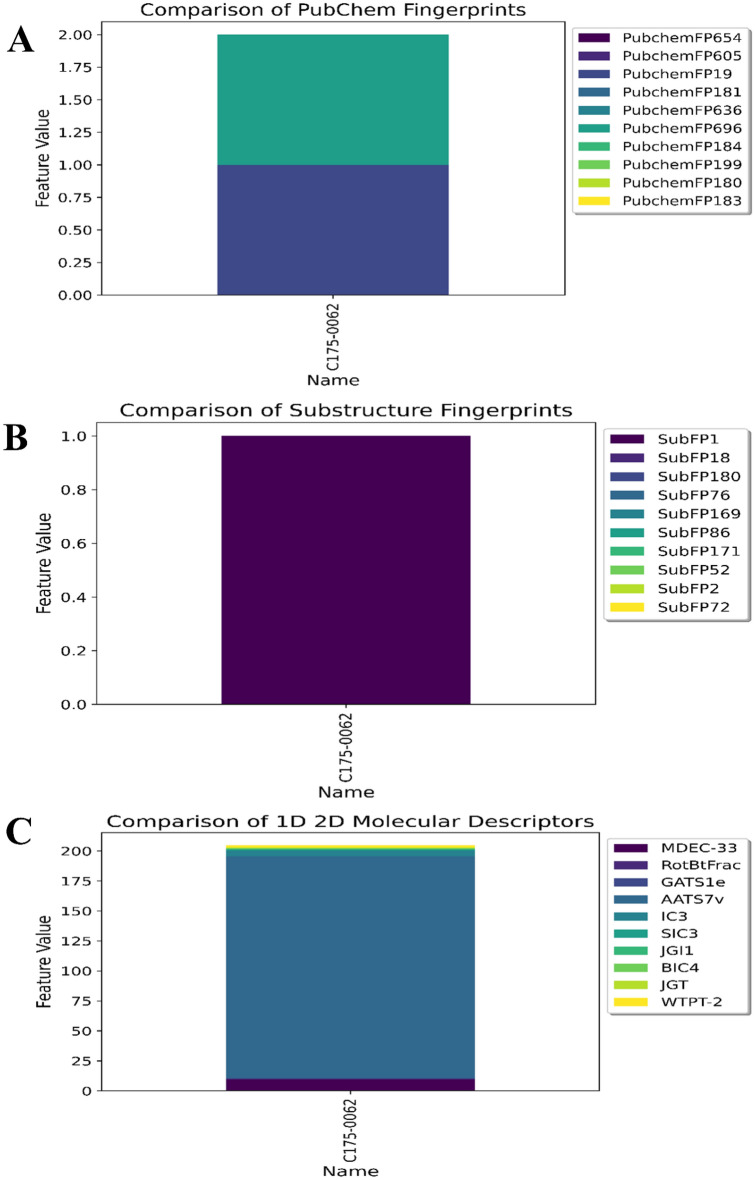
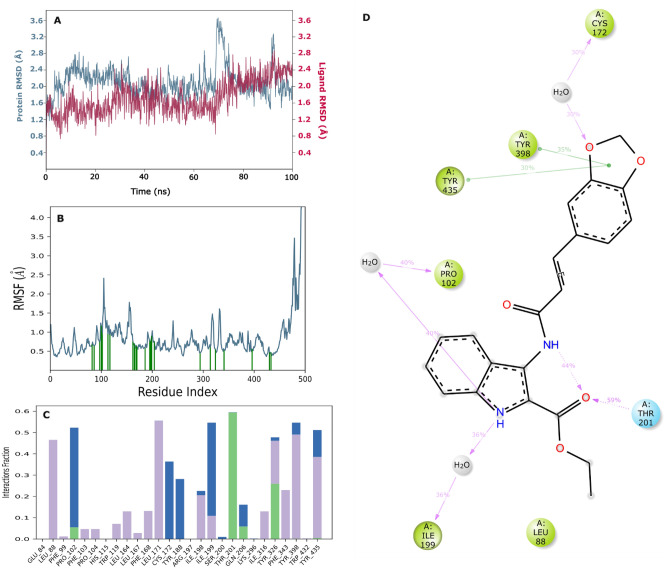
Monoamine oxidases (MAOs), specifically MAO-A and MAO-B, play important roles in the breakdown of monoamine neurotransmitters. Therefore, MAO inhibitors are crucial for treating various neurodegenerative disorders, including Parkinson's disease (PD), Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS). In this study, we developed a novel cheminformatics pipeline by generating three diverse molecular feature-based machine learning-assisted quantitative structural activity relationship (ML-QSAR) models concerning MAO-B inhibition. PubChem fingerprints, substructure fingerprints, and one-dimensional (1D) and two-dimensional (2D) molecular descriptors were implemented to unravel the structural insights responsible for decoding the origin of MAO-B inhibition in 249 non-reductant molecules. Based on a random forest ML algorithm, the final PubChem fingerprint, substructure fingerprint, and 1D and 2D molecular descriptor prediction models demonstrated significant robustness, with correlation coefficients of 0.9863, 0.9796, and 0.9852, respectively. The significant features of each predictive model responsible for MAO-B inhibition were extracted using a comprehensive variance importance plot (VIP) and correlation matrix analysis. The final predictive models were further developed as a web application, MAO-B-pred ( https://mao-b-pred.streamlit.app/ ), to allow users to predict the bioactivity of molecules against MAO-B. Molecular docking and dynamics studies were conducted to gain insight into the atomic-level molecular interactions between the ligand-receptor complexes. These findings were compared with the structural features obtained from the ML-QSAR models, which supported the mechanistic understanding of the binding phenomena. The presented models have the potential to serve as tools for identifying crucial molecular characteristics for the rational design of MAO-B target inhibitors, which may be used to develop effective drugs for neurodegenerative disorders.
单胺氧化酶(MAO),特别是 MAO-A 和 MAO-B,在单胺神经递质的分解中起着重要作用。因此,MAO 抑制剂对于治疗各种神经退行性疾病至关重要,包括帕金森病(PD)、阿尔茨海默病(AD)和肌萎缩侧索硬化症(ALS)。在这项研究中,我们通过生成三个不同的基于分子特征的机器学习辅助定量构效关系(ML-QSAR)模型来开发一种新的计算化学管道,这些模型涉及 MAO-B 抑制。我们实施了 PubChem 指纹、子结构指纹以及一维(1D)和二维(2D)分子描述符,以揭示负责解码 MAO-B 抑制起源的结构见解,这些结构涉及 249 种非还原剂分子。基于随机森林 ML 算法,最终的 PubChem 指纹、子结构指纹和 1D 和 2D 分子描述符预测模型表现出显著的稳健性,相关系数分别为 0.9863、0.9796 和 0.9852。使用综合方差重要性图(VIP)和相关矩阵分析,从每个预测模型中提取负责 MAO-B 抑制的显著特征。最终的预测模型进一步开发为一个网络应用程序,MAO-B-pred(https://mao-b-pred.streamlit.app/),以允许用户预测分子对 MAO-B 的生物活性。进行了分子对接和动力学研究,以深入了解配体-受体复合物之间的原子级分子相互作用。将这些发现与从 ML-QSAR 模型获得的结构特征进行比较,这支持了对结合现象的机制理解。所提出的模型有可能成为识别 MAO-B 靶标抑制剂合理设计的关键分子特征的工具,这可能用于开发治疗神经退行性疾病的有效药物。