Jiangxi Provincial Key Laboratory of Poultry Genetic Improvement, Nanchang Normal University, Nanchang, Jiangxi, 330032, China.
BMC Vet Res. 2024 Mar 8;20(1):93. doi: 10.1186/s12917-024-03945-9.
Bacteriophages are prokaryotic viruses that rank among the most abundant microbes in the gut but remain among the least understood, especially in quails. In this study, we surveyed the gut bacteriophage communities in 22 quails at different ages (days 20 and 70) using shotgun metagenomic sequencing. We then systematically evaluated the relationships with gut bacteria and host serum metabolites.
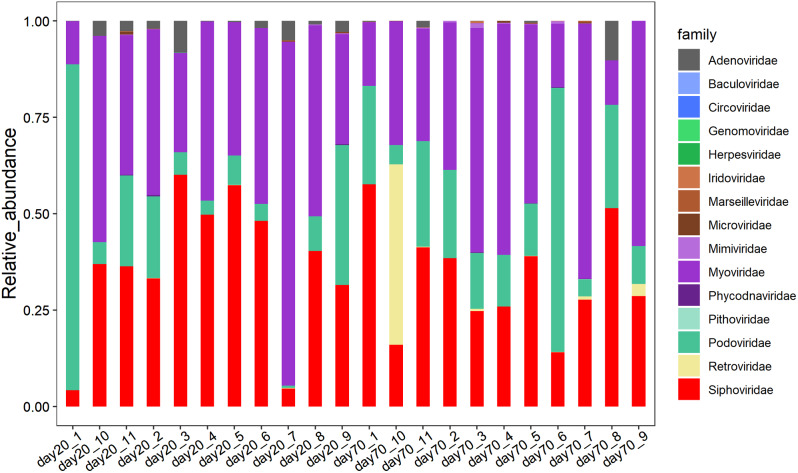
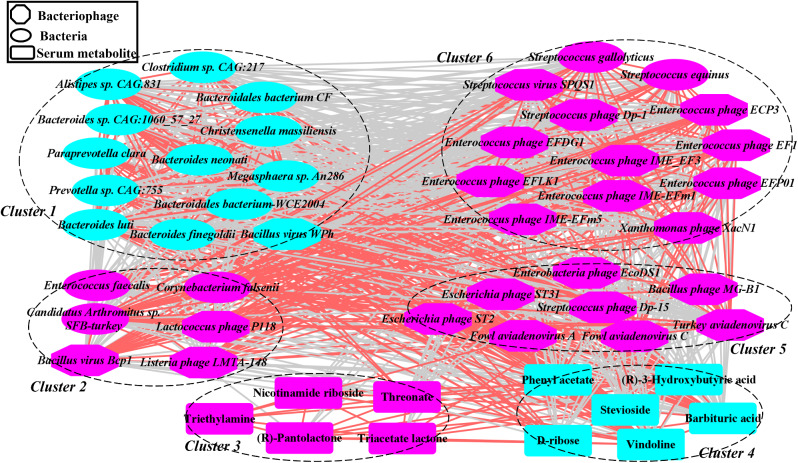
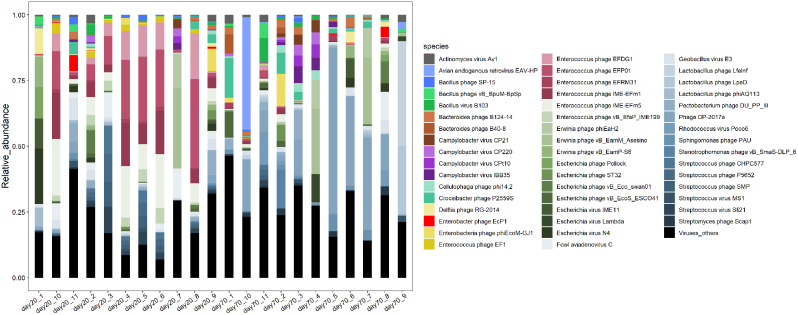
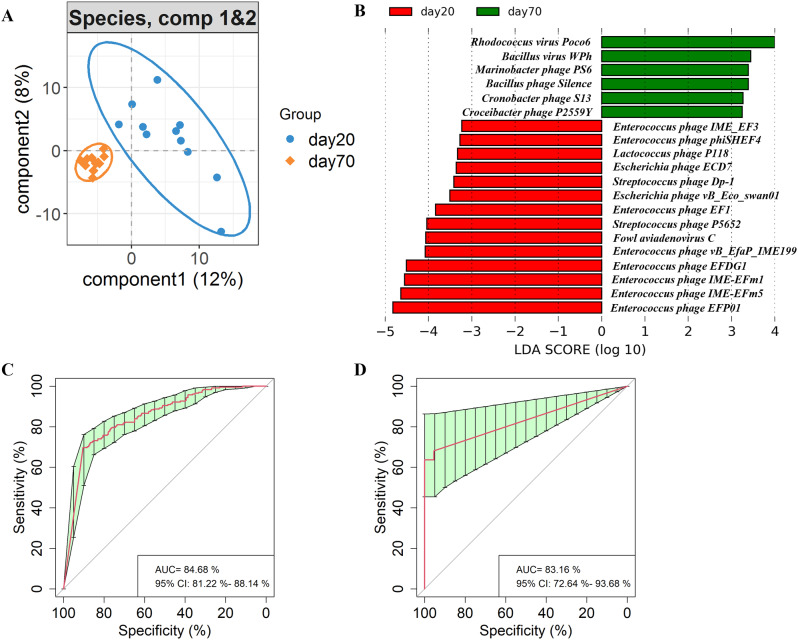
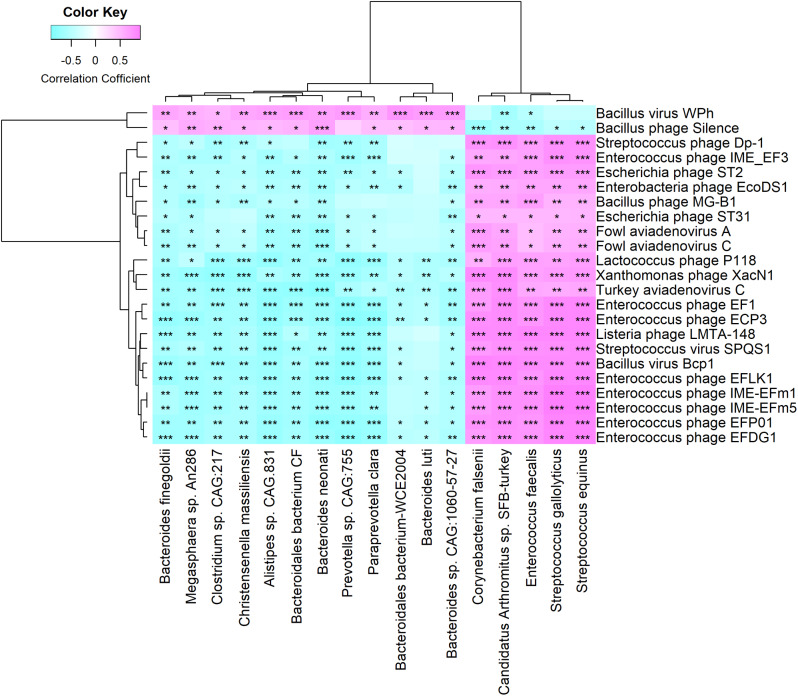
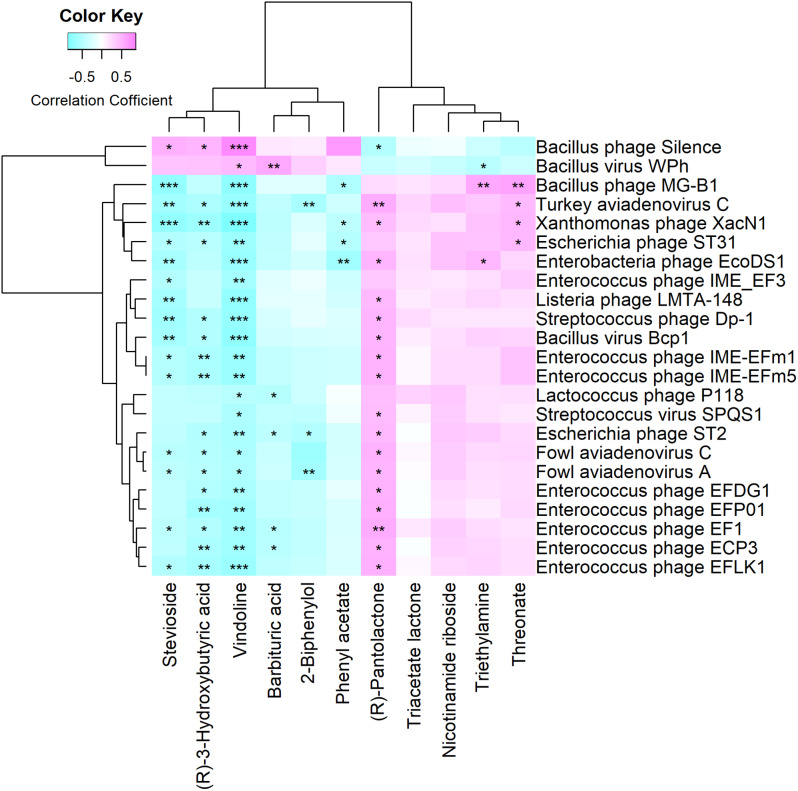
We discovered that Myoviridae and Siphoviridae were the dominant bacteriophage families in quails. Through a random forest and LEfSe analysis, we identified 23 differential bacteriophages with overlapping presence. Of these, 21 bacteriophages (e.g., Enterococcus phage IME-EFm5 and Enterococcus phage IME-EFm1) showed higher abundances in the day 20 group, while two bacteriophages (Bacillus phage Silence and Bacillus virus WPh) were enriched in the day 70 group. These key bacteriophages can serve as biomarkers for quail sexual maturity. Additionally, the differential bacteriophages significantly correlated with specific bacterial species and shifts in the functional capacities of the gut microbiome. For example, Enterococcus phages (e.g., Enterococcus phage EFP01, Enterococcus phage IME-EFm5, and Enterococcus phage IME-EFm1) were significantly (P < 0.001, FDR) and positively correlated with Enterococcus faecalis. However, the relationships between the host serum metabolites and either bacteriophages or bacterial species varied. None of the bacteriophages significantly (P > 0.05, FDR) correlated with nicotinamide riboside and triacetate lactone. In contrast, some differential bacterial species (e.g., Christensenella massiliensis and Bacteroides neonati) significantly (P < 0.05, FDR) correlated with nicotinamide riboside and triacetate lactone. Furthermore, characteristic successional alterations in gut bacteriophages, bacteria, and host serum metabolites across different ages highlighted a sexual maturity transition coexpression network.
This study improves our understanding of the gut bacteriophage characteristics in quails and offers profound insights into the interactions among gut bacteriophages, bacteria, and host serum metabolites during the quail's sexual maturity transition.
噬菌体是原核病毒,在肠道中数量最多的微生物之一,但了解甚少,尤其是在鹌鹑中。在这项研究中,我们使用鸟枪法宏基因组测序对 22 只不同日龄(20 天和 70 天)的鹌鹑的肠道噬菌体群落进行了调查。然后,我们系统地评估了与肠道细菌和宿主血清代谢物的关系。
我们发现,肌尾噬菌体科和长尾噬菌体科是鹌鹑中主要的噬菌体科。通过随机森林和 Lefe 分析,我们鉴定出 23 个具有重叠存在的差异噬菌体。其中,21 个噬菌体(如肠球菌噬菌体 IME-EFm5 和肠球菌噬菌体 IME-EFm1)在 20 天组中的丰度较高,而两个噬菌体(芽孢杆菌噬菌体 Silence 和芽孢杆菌病毒 WPh)在 70 天组中富集。这些关键噬菌体可以作为鹌鹑性成熟的生物标志物。此外,差异噬菌体与特定细菌物种和肠道微生物组功能能力的变化显著相关。例如,肠球菌噬菌体(如肠球菌噬菌体 EFP01、肠球菌噬菌体 IME-EFm5 和肠球菌噬菌体 IME-EFm1)与粪肠球菌呈显著(P<0.001, FDR)正相关。然而,宿主血清代谢物与噬菌体或细菌之间的关系各不相同。没有一种噬菌体与烟酰胺核糖苷和三醋酸盐显著相关(P>0.05, FDR)。相反,一些差异细菌物种(如马西利昂氏菌和新生儿拟杆菌)与烟酰胺核糖苷和三醋酸盐显著相关(P<0.05, FDR)。此外,不同年龄肠道噬菌体、细菌和宿主血清代谢物的特征性连续变化突出了性成熟过渡的共表达网络。
本研究提高了我们对鹌鹑肠道噬菌体特征的认识,并深入了解了鹌鹑性成熟过渡期间肠道噬菌体、细菌和宿主血清代谢物之间的相互作用。