Pascariu Matei, Bernasconi Leonardo, Krzystyniak Matthew, Taylor James, Rudić Svemir
ISIS Neutron and Muon Source, Rutherford Appleton Laboratory, STFC, Harwell Campus, Chilton, Oxfordshire OX11 0QX, U.K.
Department of Chemistry, The University of Manchester, Oxford Road, Manchester M13 9PL, U.K.
J Phys Chem A. 2024 Mar 21;128(11):2111-2120. doi: 10.1021/acs.jpca.4c00266. Epub 2024 Mar 12.
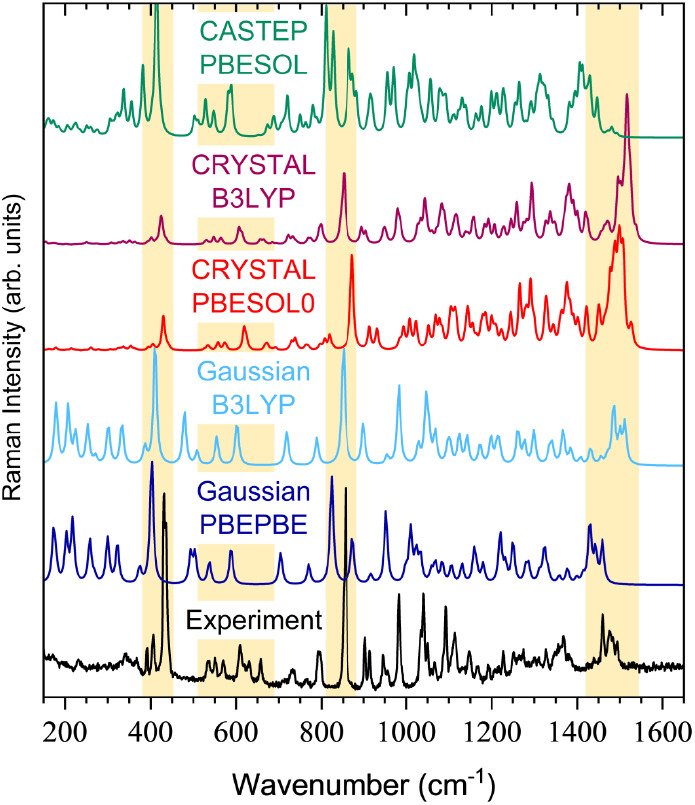
This study presents a comprehensive analysis of the vibrational spectra of methyl-β-D-ribofuranoside. Employing a combination of inelastic neutron scattering, Raman, and infrared spectroscopy allows for the observation of all modes regardless of the selection rules. The experimental techniques were complemented by density functional theory computational methods using both gas-phase () and solid-state (, ) approaches to provide an unambiguous assignment of the defining vibrational features. Two distinct structures of the molecule were identified in the unit cell, differentiated mainly by the orientation of the furanose ring O-H bonds. The low-energy region of the spectrum (<400 cm) is dominated by lattice vibrations and functional group rotation, while the midenergy region is dominated by out-of-plane bending motions of the furanose ring (400-900 cm) and by C-H bending in the methyl and methylene groups (1400-1600 cm). The high-energy region (>2800 cm) encompasses the C-H and O-H stretching modes and offers convincing evidence of at least one H-bonding interaction between the two structures of methyl-β-D-ribofuranoside.
本研究对甲基-β-D-呋喃核糖苷的振动光谱进行了全面分析。采用非弹性中子散射、拉曼光谱和红外光谱相结合的方法,无论选择规则如何,都能观察到所有模式。实验技术通过使用气相()和固态(,)方法的密度泛函理论计算方法进行补充,以明确确定主要振动特征。在晶胞中鉴定出该分子的两种不同结构,主要区别在于呋喃糖环O-H键的取向。光谱的低能量区域(<400 cm)主要由晶格振动和官能团旋转主导,而中能量区域主要由呋喃糖环的面外弯曲运动(400 - 900 cm)以及甲基和亚甲基中的C-H弯曲(1400 - 1600 cm)主导。高能量区域(>2800 cm)包含C-H和O-H伸缩模式,并提供了甲基-β-D-呋喃核糖苷两种结构之间至少存在一种氢键相互作用的有力证据。