School of Physics, Huazhong University of Science and Technology, Wuhan, China.
Elife. 2024 Apr 2;12:RP92184. doi: 10.7554/eLife.92184.
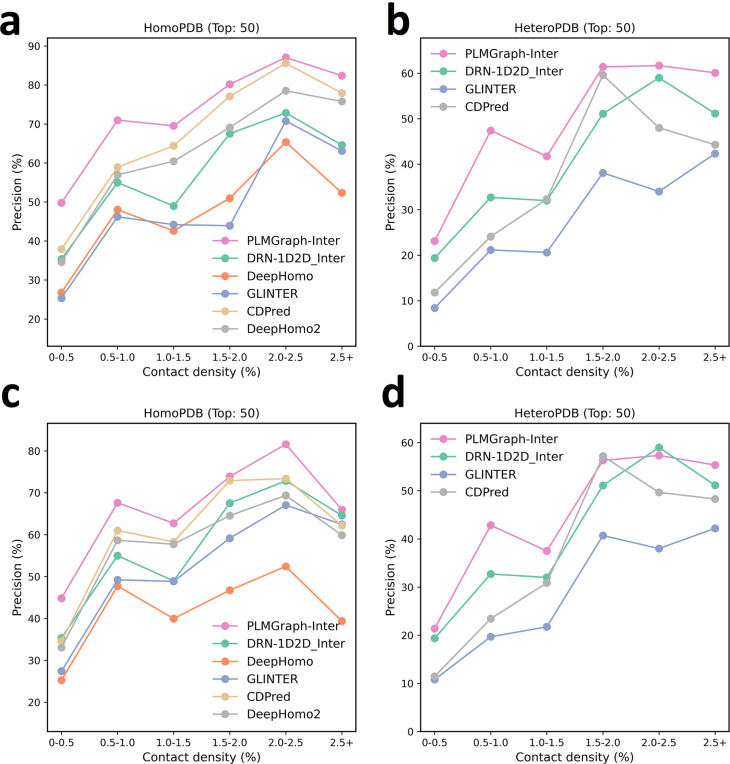
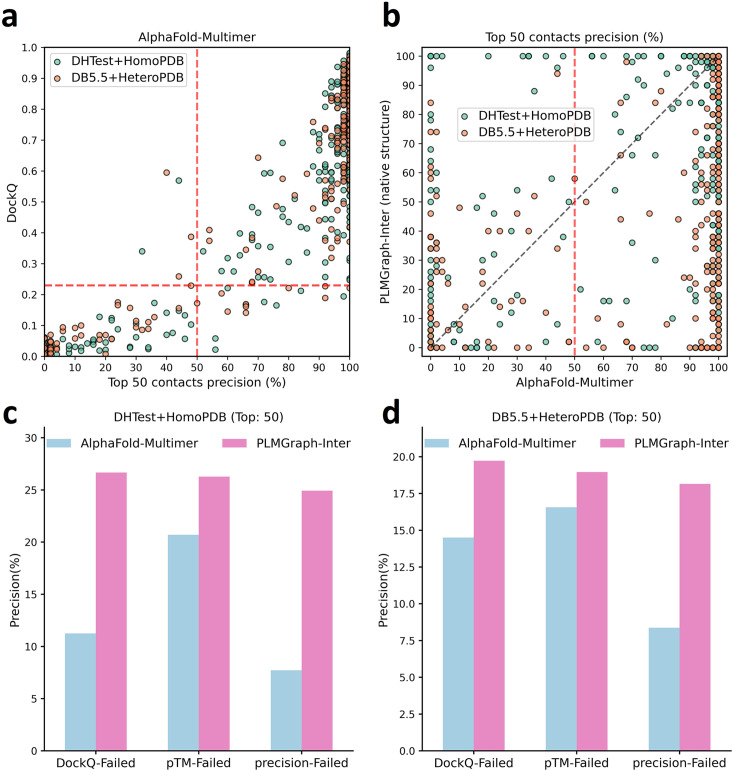
Accurate prediction of contacting residue pairs between interacting proteins is very useful for structural characterization of protein-protein interactions. Although significant improvement has been made in inter-protein contact prediction recently, there is still a large room for improving the prediction accuracy. Here we present a new deep learning method referred to as PLMGraph-Inter for inter-protein contact prediction. Specifically, we employ rotationally and translationally invariant geometric graphs obtained from structures of interacting proteins to integrate multiple protein language models, which are successively transformed by graph encoders formed by geometric vector perceptrons and residual networks formed by dimensional hybrid residual blocks to predict inter-protein contacts. Extensive evaluation on multiple test sets illustrates that PLMGraph-Inter outperforms five top inter-protein contact prediction methods, including DeepHomo, GLINTER, CDPred, DeepHomo2, and DRN-1D2D_Inter, by large margins. In addition, we also show that the prediction of PLMGraph-Inter can complement the result of AlphaFold-Multimer. Finally, we show leveraging the contacts predicted by PLMGraph-Inter as constraints for protein-protein docking can dramatically improve its performance for protein complex structure prediction.
准确预测相互作用蛋白质之间的接触残基对蛋白质-蛋白质相互作用的结构特征化非常有用。尽管最近在蛋白质间接触预测方面取得了重大进展,但仍有很大的提高预测准确性的空间。在这里,我们提出了一种新的深度学习方法,称为 PLMGraph-Inter,用于蛋白质间接触预测。具体来说,我们利用来自相互作用蛋白质结构的旋转和平移不变的几何图形来整合多个蛋白质语言模型,这些模型依次由几何向量感知机和维度混合残差块组成的残差网络形成的图编码器进行转换,以预测蛋白质间的接触。在多个测试集上的广泛评估表明,PLMGraph-Inter 优于包括 DeepHomo、GLINTER、CDPred、DeepHomo2 和 DRN-1D2D_Inter 在内的五种顶级蛋白质间接触预测方法,具有很大的优势。此外,我们还表明,PLMGraph-Inter 的预测可以补充 AlphaFold-Multimer 的结果。最后,我们表明,利用 PLMGraph-Inter 预测的接触作为蛋白质-蛋白质对接的约束条件可以显著提高其用于蛋白质复合物结构预测的性能。