You Wenting, Knoops Kèvin, Berendschot Tos T J M, Benedikter Birke J, Webers Carroll A B, Reutelingsperger Chris P M, Gorgels Theo G M F
University Eye Clinic Maastricht UMC+, Maastricht University Medical Center+, Maastricht, The Netherlands.
Department of Biochemistry, CARIM School for Cardiovascular Disease, Maastricht University, Maastricht, The Netherlands.
Cell Death Discov. 2024 Apr 17;10(1):180. doi: 10.1038/s41420-024-01953-0.
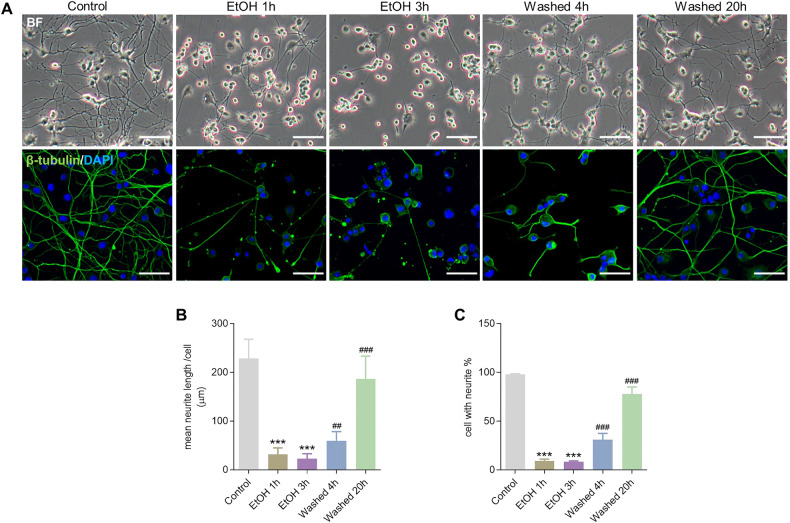
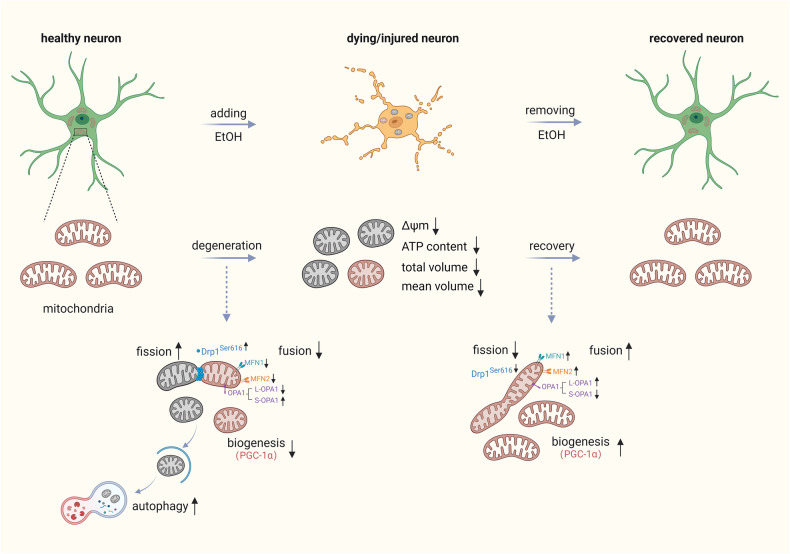
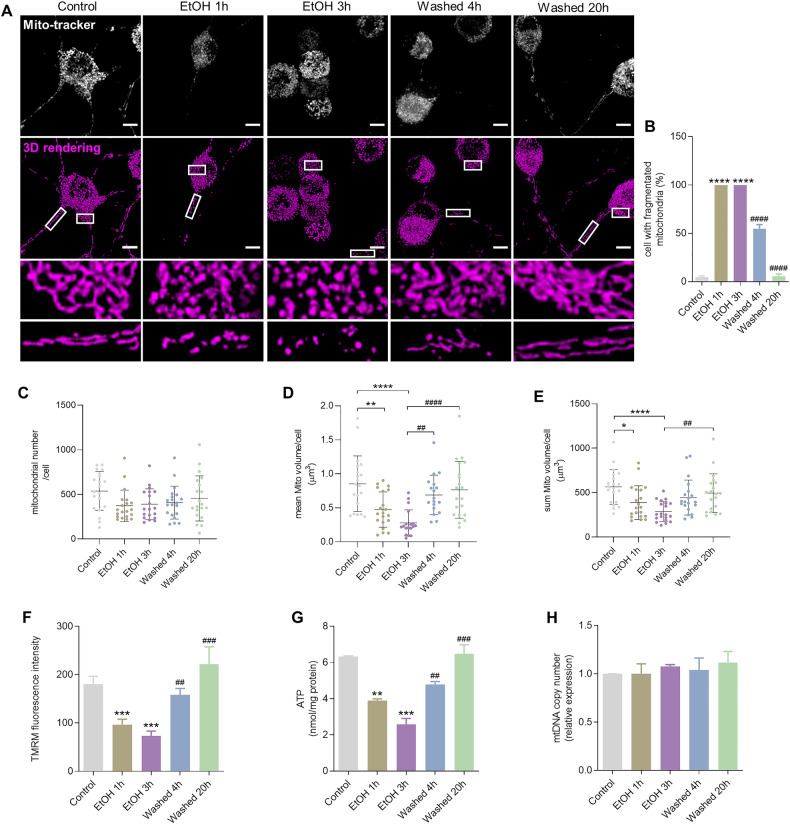
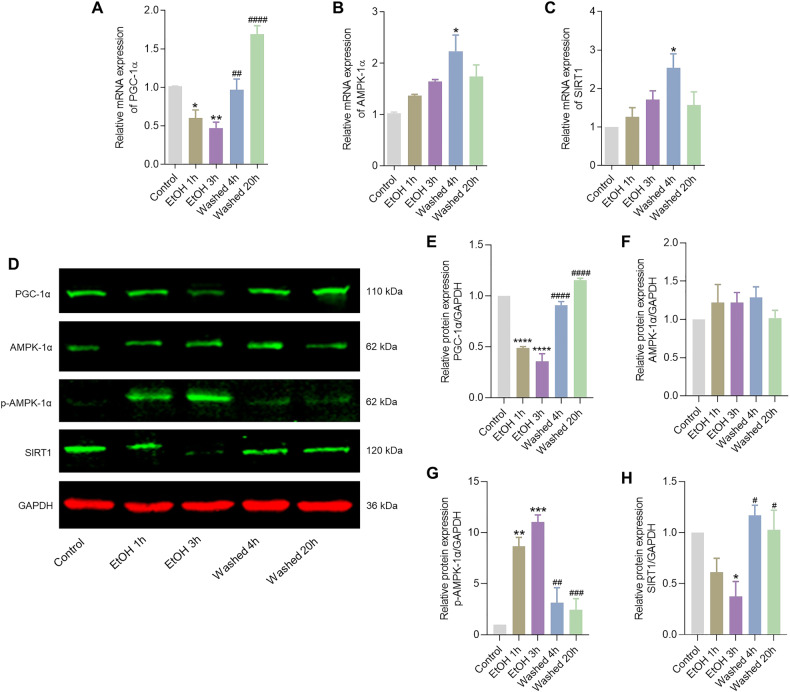
Neurodegenerative disorders are characterized by the progressive loss of structure and function of neurons, often including the death of the neuron. Previously, we reported that, by removing the cell death stimulus, dying/injured neurons could survive and recover from the process of regulated cell death, even if the cells already displayed various signs of cellular damage. Now we investigated the role of mitochondrial dynamics (fission/fusion, biogenesis, mitophagy) in both degeneration and in recovery of neuronal cells. In neuronal PC12 cells, exposure to ethanol (EtOH) induced massive neurite loss along with widespread mitochondrial fragmentation, mitochondrial membrane potential loss, reduced ATP production, and decreased total mitochondrial volume. By removing EtOH timely all these mitochondrial parameters recovered to normal levels. Meanwhile, cells regrew neurites and survived. Study of the mitochondrial dynamics showed that autophagy was activated only during the cellular degeneration phase (EtOH treatment) but not in the recovery phase (EtOH removed), and it was not dependent on the Parkin/PINK1 mediated mitophagy pathway. Protein expression of key regulators of mitochondrial fission, phospho-Drp1 and S-OPA1, increased during EtOH treatment and recovered to normal levels after removing EtOH. In addition, the critical role of PGC-1α mediated mitochondrial biogenesis in cellular recovery was revealed: inhibition of PGC-1α using SR-18292 after EtOH removal significantly impeded recovery of mitochondrial damage, regeneration of neurites, and cell survival in a concentration-dependent manner. Taken together, our study showed reversibility of mitochondrial morphological and functional damage in stressed neuronal cells and revealed that PGC-1α mediated mitochondrial biogenesis played a critical role in the cellular recovery. This molecular mechanism could be a target for neuroprotection and neurorescue in neurodegenerative diseases.
神经退行性疾病的特征是神经元结构和功能的逐渐丧失,通常包括神经元死亡。此前,我们报道过,通过去除细胞死亡刺激因素,濒死/受损的神经元能够存活下来,并从程序性细胞死亡过程中恢复,即便这些细胞已经表现出各种细胞损伤迹象。现在我们研究了线粒体动力学(裂变/融合、生物合成、线粒体自噬)在神经元细胞退化和恢复过程中的作用。在神经元PC12细胞中,暴露于乙醇(EtOH)会导致大量神经突丧失,同时伴有广泛的线粒体碎片化、线粒体膜电位丧失、ATP生成减少以及线粒体总体积减小。通过及时去除EtOH,所有这些线粒体参数都恢复到了正常水平。与此同时,细胞重新长出神经突并存活下来。对线粒体动力学的研究表明,自噬仅在细胞退化阶段(EtOH处理期间)被激活,而在恢复阶段(EtOH去除后)未被激活,且其不依赖于Parkin/PINK1介导的线粒体自噬途径。线粒体裂变的关键调节因子磷酸化动力蛋白1(phospho-Drp1)和S-视神经萎缩蛋白1(S-OPA1)的蛋白表达在EtOH处理期间增加,并在去除EtOH后恢复到正常水平。此外,还揭示了过氧化物酶体增殖物激活受体γ共激活因子1α(PGC-1α)介导的线粒体生物合成在细胞恢复中的关键作用:在去除EtOH后使用SR-18292抑制PGC-1α会以浓度依赖的方式显著阻碍线粒体损伤的恢复、神经突的再生以及细胞存活。综上所述,我们的研究表明应激神经元细胞中线粒体形态和功能损伤具有可逆性,并揭示了PGC-1α介导的线粒体生物合成在细胞恢复中起着关键作用。这种分子机制可能成为神经退行性疾病神经保护和神经挽救的靶点。