Department of Computer Science, University of Oxford, Oxford, UK.
Oxford Centre for Clinical Magnetic Resonance Research (OCMR), Radcliffe Department of Medicine, Division of Cardiovascular Medicine, University of Oxford, Oxford, UK.
Cardiovasc Res. 2024 Jul 2;120(8):914-926. doi: 10.1093/cvr/cvae086.
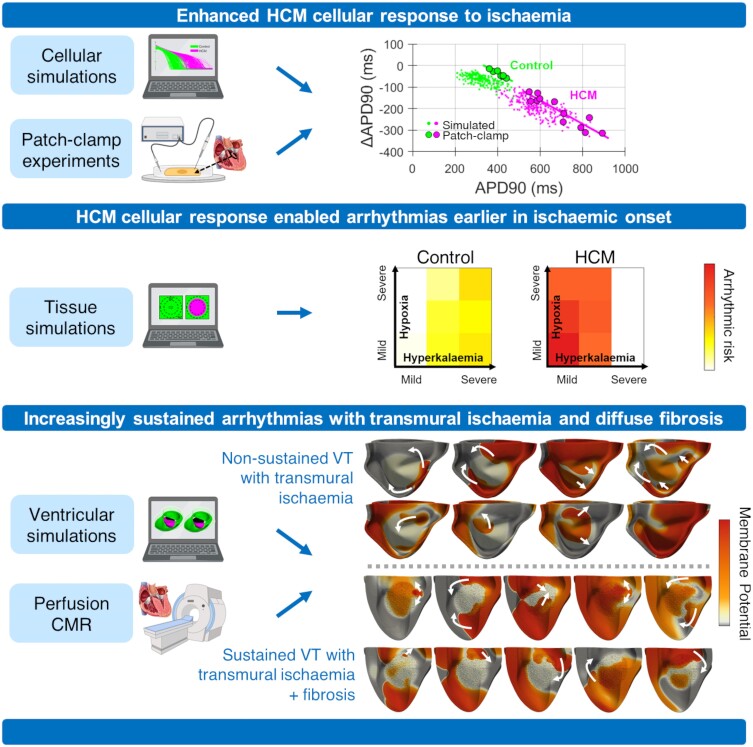
Lethal arrhythmias in hypertrophic cardiomyopathy (HCM) are widely attributed to myocardial ischaemia and fibrosis. How these factors modulate arrhythmic risk remains largely unknown, especially as invasive mapping protocols are not routinely used in these patients. By leveraging multiscale digital twin technologies, we aim to investigate ischaemic mechanisms of increased arrhythmic risk in HCM.
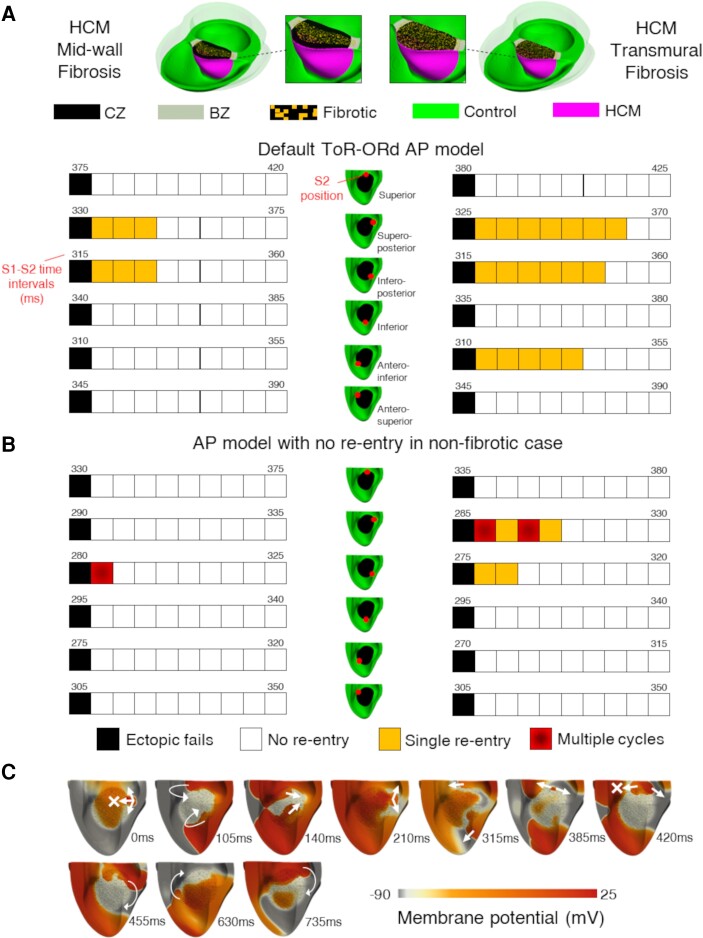
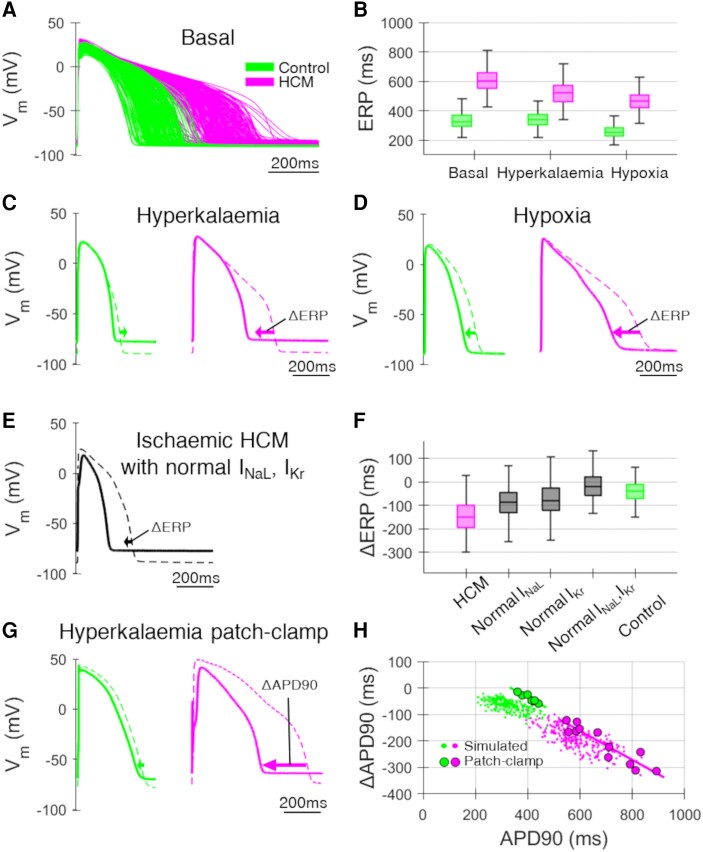
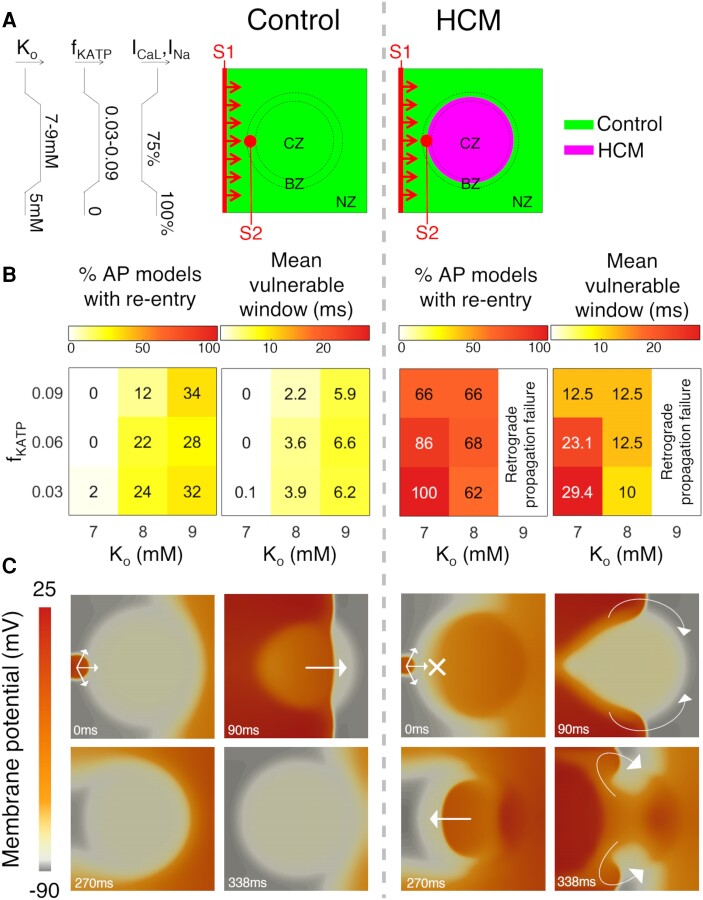
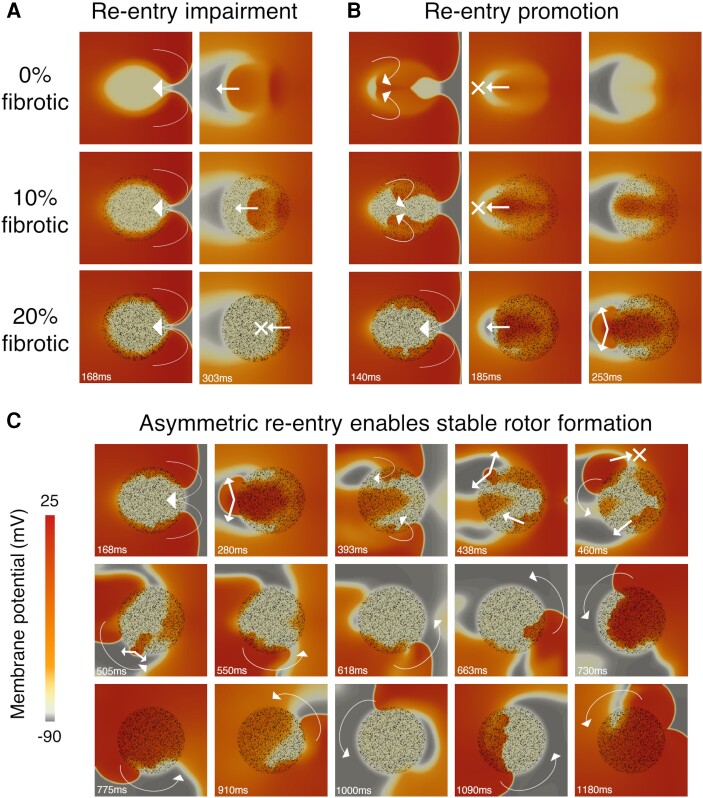
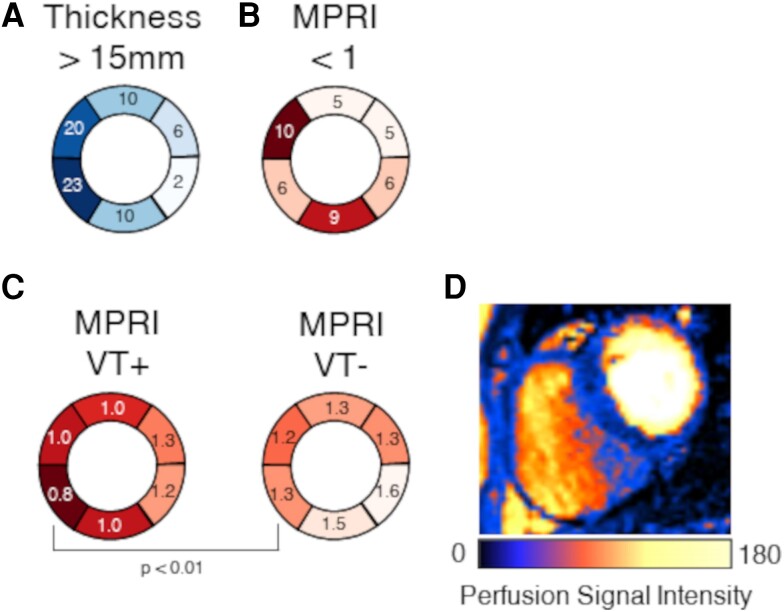
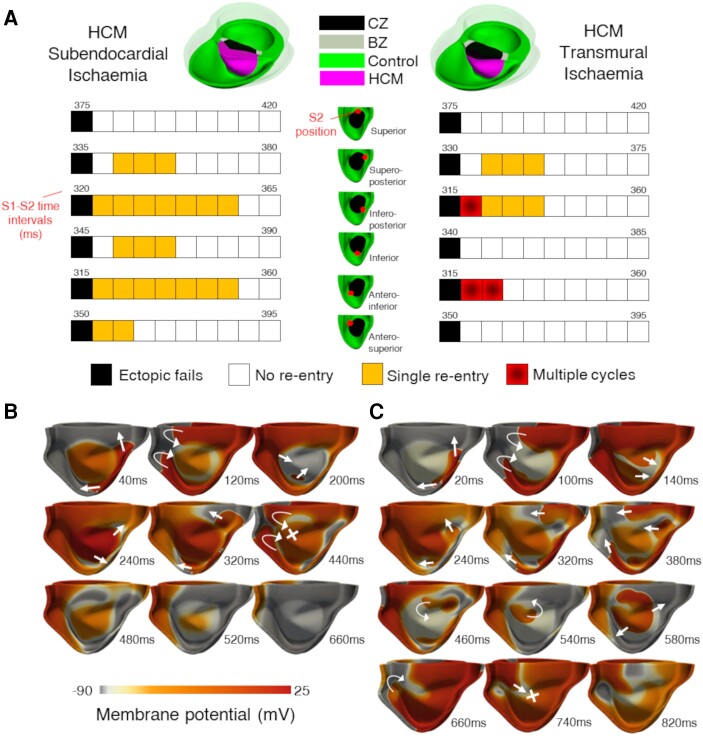
Computational models of human HCM cardiomyocytes, tissue, and ventricles were used to simulate outcomes of Phase 1A acute myocardial ischaemia. Cellular response predictions were validated with patch-clamp studies of human HCM cardiomyocytes (n = 12 cells, N = 5 patients). Ventricular simulations were informed by typical distributions of subendocardial/transmural ischaemia as analysed in perfusion scans (N = 28 patients). S1-S2 pacing protocols were used to quantify arrhythmic risk for scenarios in which regions of septal obstructive hypertrophy were affected by (i) ischaemia, (ii) ischaemia and impaired repolarization, and (iii) ischaemia, impaired repolarization, and diffuse fibrosis. HCM cardiomyocytes exhibited enhanced action potential and abnormal effective refractory period shortening to ischaemic insults. Analysis of ∼75 000 re-entry induction cases revealed that the abnormal HCM cellular response enabled establishment of arrhythmia at milder ischaemia than otherwise possible in healthy myocardium, due to larger refractoriness gradients that promoted conduction block. Arrhythmias were more easily sustained in transmural than subendocardial ischaemia. Mechanisms of ischaemia-fibrosis interaction were strongly electrophysiology dependent. Fibrosis enabled asymmetric re-entry patterns and break-up into sustained ventricular tachycardia.
HCM ventricles exhibited an increased risk to non-sustained and sustained re-entry, largely dominated by an impaired cellular response and deleterious interactions with the diffuse fibrotic substrate.
肥厚型心肌病(HCM)中的致死性心律失常广泛归因于心肌缺血和纤维化。这些因素如何调节心律失常风险在很大程度上仍然未知,特别是因为在这些患者中通常不使用侵入性映射方案。通过利用多尺度数字孪生技术,我们旨在研究 HCM 中增加心律失常风险的缺血机制。
使用人类 HCM 心肌细胞、组织和心室的计算模型来模拟 1A 期急性心肌缺血的结果。通过对人类 HCM 心肌细胞(n = 12 个细胞,N = 5 个患者)的膜片钳研究验证了细胞反应预测。心室模拟基于灌注扫描中分析的心内膜/中层缺血的典型分布(N = 28 个患者)。S1-S2 起搏方案用于量化当间隔阻塞性肥厚区域受到以下情况影响时的心律失常风险:(i)缺血,(ii)缺血和复极化受损,以及(iii)缺血、复极化受损和弥漫性纤维化。HCM 心肌细胞对缺血性损伤表现出增强的动作电位和异常的有效不应期缩短。对约 75000 个折返诱导病例的分析表明,由于较大的不应期梯度促进传导阻滞,异常的 HCM 细胞反应使心律失常在比健康心肌更轻微的缺血条件下得以建立,这是因为正常心肌不可能建立心律失常。与心内膜下缺血相比,透壁性缺血更容易维持心律失常。缺血-纤维化相互作用的机制强烈依赖于电生理学。纤维化使非对称折返模式和转化为持续室性心动过速成为可能。
HCM 心室表现出非持续和持续折返的风险增加,主要由细胞反应受损和与弥漫性纤维化基质的有害相互作用引起。