Shen Wangzhen, Nwosu Gerald, Honer Michael, Clasadonte Jerome, Schmalzbauer Svenja, Biven Marshall, Langer Katherine, Flamm Carson, Poliquin Sarah, Mermer Felicia, Dedeurwaerdere Stefanie, Hernandez Maria-Clemencia, Kang Jing-Qiong
Department of Neurology, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Neuroscience Graduate Program, Vanderbilt University, Nashville, TN 37232, USA.
Brain Commun. 2024 Apr 22;6(2):fcae110. doi: 10.1093/braincomms/fcae110. eCollection 2024.
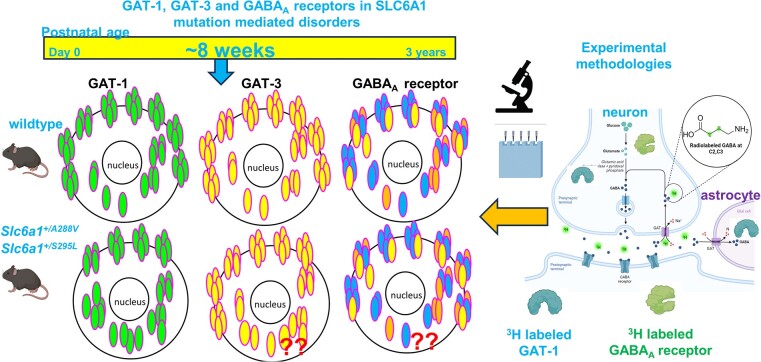
We have previously characterized the molecular mechanisms for variants in γ-aminobutyric acid transporter 1-encoding solute carrier family 6-member 1 () and concluded that a partial or complete loss of γ-aminobutyric acid uptake due to impaired protein trafficking is the primary aetiology. Impairment of γ-aminobutyric acid transporter 1 function could cause compensatory changes in the expression of γ-aminobutyric acid receptors, which, in turn, modify disease pathophysiology and phenotype. Here we used different approaches including radioactive H γ-aminobutyric acid uptake in cells and synaptosomes, immunohistochemistry and confocal microscopy as well as brain slice surface protein biotinylation to characterize and mice, representative of a partial or a complete loss of function of mutations, respectively. We employed the γ-aminobutyric acid transporter 1-specific inhibitor [H]tiagabine binding and GABA receptor subunit-specific radioligand binding to profile the γ-aminobutyric acid transporter 1 and GABA receptor expression in major brain regions such as cortex, cerebellum, hippocampus and thalamus. We also determined the total and surface expression of γ-aminobutyric acid transporter 1, γ-aminobutyric acid transporter 3 and expression of GABA receptor in the major brain regions in the knockin mice. We found that γ-aminobutyric acid transporter 1 protein was markedly reduced in cortex, hippocampus, thalamus and cerebellum in both mutant mouse lines. Consistent with the findings of reduced γ-aminobutyric acid uptake for both γ-aminobutyric acid transporter 1(A288V) and γ-aminobutyric acid transporter 1(S295L), both the total and the γ-aminobutyric acid transporter 1-mediated H γ-aminobutyric acid reuptake was reduced. We found that γ-aminobutyric acid transporter 3 is only abundantly expressed in the thalamus and there was no compensatory increase of γ-aminobutyric acid transporter 3 in either of the mutant mouse lines. γ-Aminobutyric acid transporter 1 was reduced in both somatic regions and nonsomatic regions in both mouse models, in which a ring-like structure was identified only in the mouse, suggesting more γ-aminobutyric acid transporter 1 retention inside endoplasmic reticulum in the mouse. The [H]tiagabine binding was similar in both mouse models despite the difference in γ-aminobutyric acid uptake function and γ-aminobutyric acid transporter 1 protein expression for both mutations. There were no differences in GABA receptor subtype expression, except for a small increase in the expression of α5 subunits of GABA receptor in the hippocampus of homozygous mice, suggesting a potential interaction between the expression of this GABA receptor subtype and the mutant γ-aminobutyric acid transporter 1. The study provides the first comprehensive characterization of the mutations in two representative mouse models. Because both γ-aminobutyric acid transporter 1 and GABA receptors are targets for anti-seizure medications, the findings from this study can help guide tailored treatment options based on the expression and function of γ-aminobutyric acid transporter 1 and GABA receptor in mutation-mediated neurodevelopmental and epileptic encephalopathies.
我们之前已对编码γ-氨基丁酸转运体1的溶质载体家族6成员1()中的变异体的分子机制进行了表征,并得出结论,由于蛋白质运输受损导致γ-氨基丁酸摄取部分或完全丧失是主要病因。γ-氨基丁酸转运体1功能受损可能会导致γ-氨基丁酸受体表达的代偿性变化,进而改变疾病的病理生理学和表型。在此,我们采用了不同方法,包括在细胞和突触体中进行放射性Hγ-氨基丁酸摄取、免疫组织化学和共聚焦显微镜检查以及脑片表面蛋白生物素化,以分别表征和小鼠,它们分别代表突变功能部分丧失或完全丧失的典型情况。我们使用γ-氨基丁酸转运体1特异性抑制剂[H]噻加宾结合以及GABA受体亚基特异性放射性配体结合,来分析主要脑区如皮质、小脑、海马体和丘脑的γ-氨基丁酸转运体1和GABA受体表达情况。我们还测定了敲入小鼠主要脑区中γ-氨基丁酸转运体1、γ-氨基丁酸转运体3的总表达和表面表达以及GABA受体的表达。我们发现,在两个突变小鼠品系中,皮质、海马体、丘脑和小脑中的γ-氨基丁酸转运体1蛋白均显著减少。与γ-氨基丁酸转运体1(A288V)和γ-氨基丁酸转运体1(S295L)的γ-氨基丁酸摄取减少的结果一致,γ-氨基丁酸转运体1介导的Hγ-氨基丁酸再摄取总量均减少。我们发现γ-氨基丁酸转运体3仅在丘脑中大量表达,且在任何一个突变小鼠品系中γ-氨基丁酸转运体3均无代偿性增加。在两种小鼠模型中,γ-氨基丁酸转运体1在躯体区域和非躯体区域均减少,其中仅在小鼠中鉴定出一种环状结构,这表明小鼠内质网中保留了更多的γ-氨基丁酸转运体1。尽管两种突变的γ-氨基丁酸摄取功能和γ-氨基丁酸转运体1蛋白表达存在差异,但两种小鼠模型中的[H]噻加宾结合情况相似。除了纯合小鼠海马体中GABA受体α5亚基表达略有增加外,GABA受体亚型表达没有差异,这表明该GABA受体亚型的表达与突变的γ-氨基丁酸转运体1之间可能存在相互作用。该研究首次全面表征了两种代表性小鼠模型中的突变情况。由于γ-氨基丁酸转运体1和GABA受体均为抗癫痫药物的靶点,本研究结果有助于基于γ-氨基丁酸转运体1和GABA受体在突变介导的神经发育和癫痫性脑病中的表达和功能来指导个性化治疗方案的选择。