Department of Pharmaceutical Chemistry, Manipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education, Manipal, Karnataka, India, 576104.
Department of Medical Affairs, Curie Sciences Private Limited, Samastipur, Bihar, India, 848125.
Sci Rep. 2024 May 17;14(1):11315. doi: 10.1038/s41598-024-61901-x.
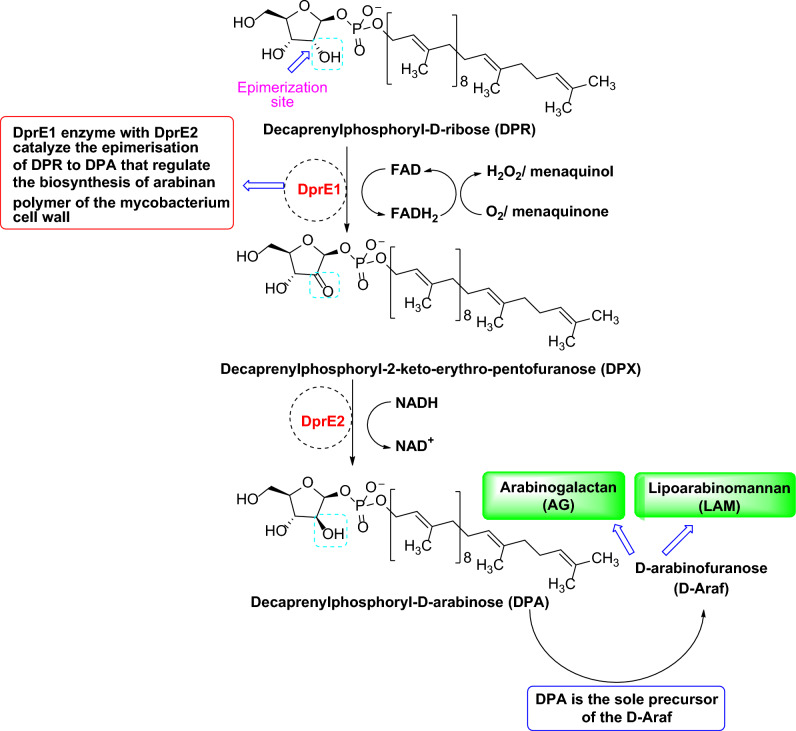
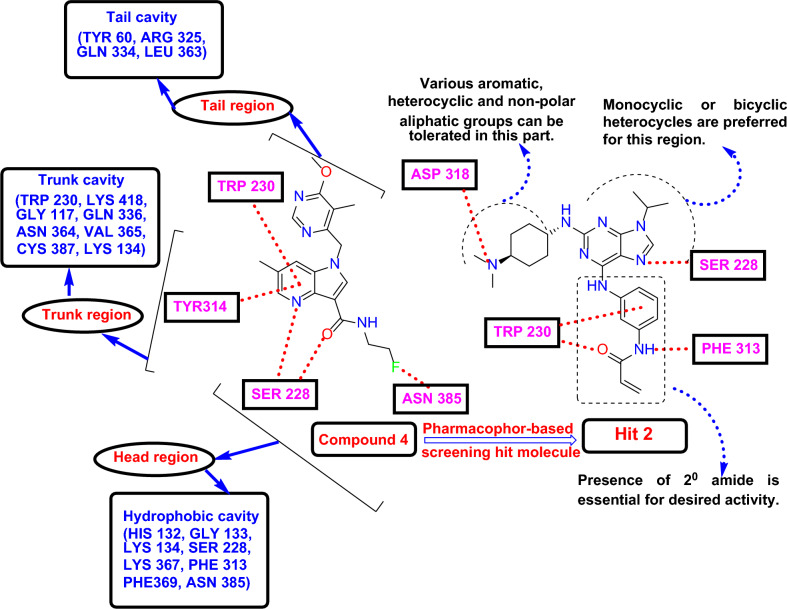
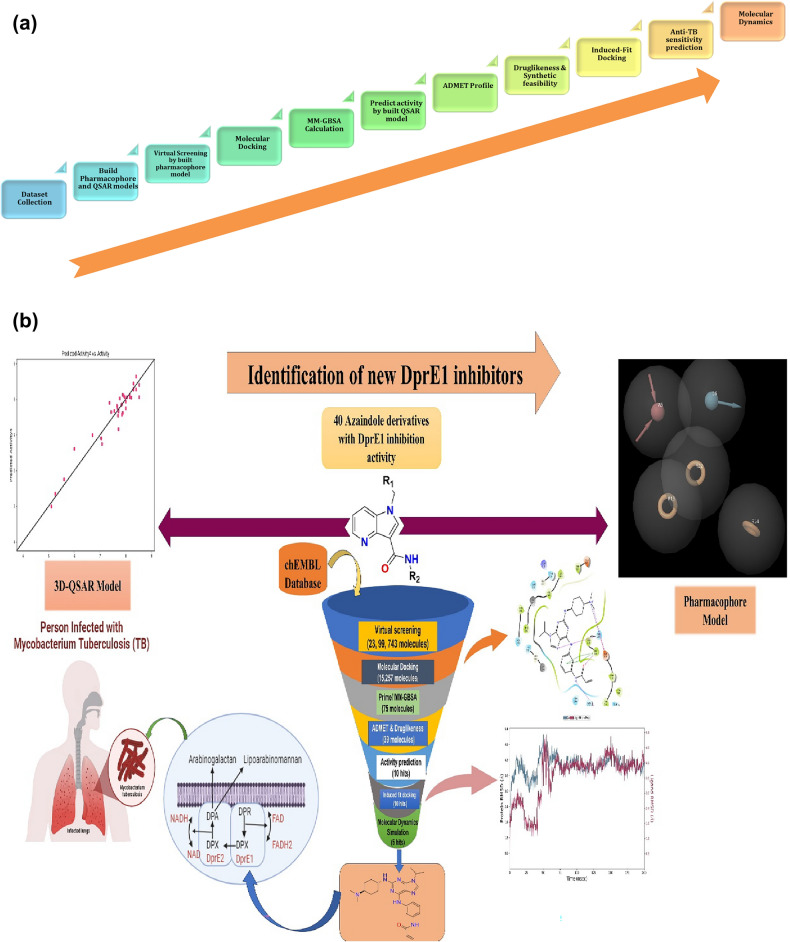
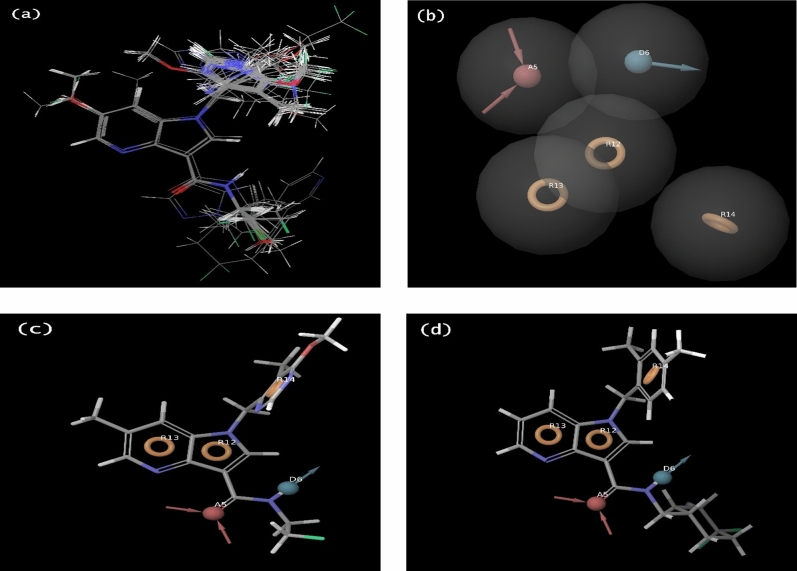
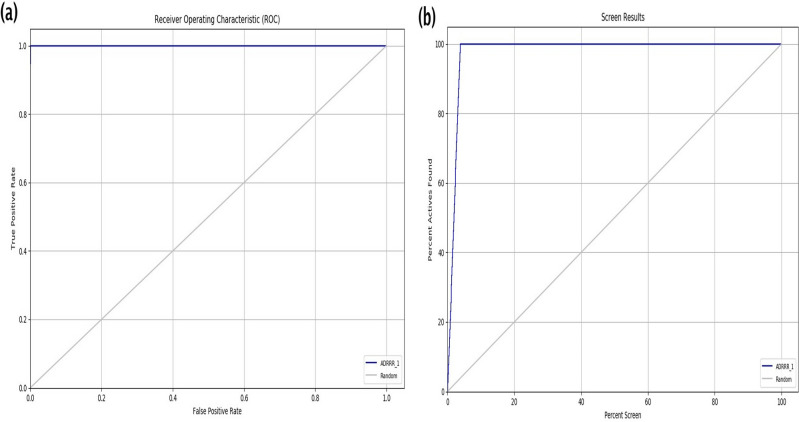
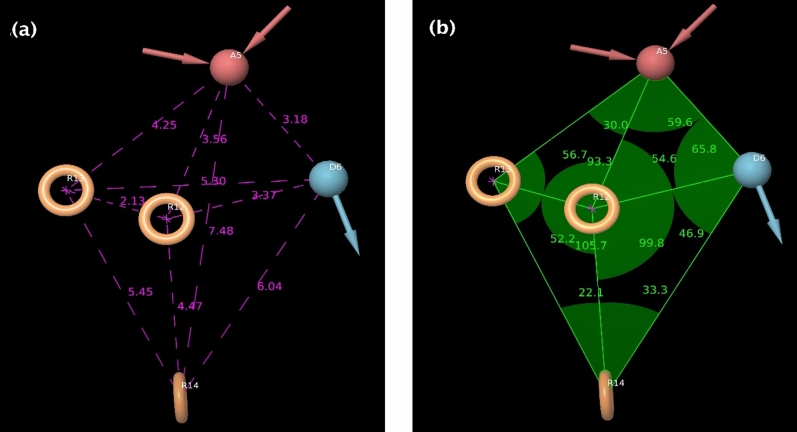
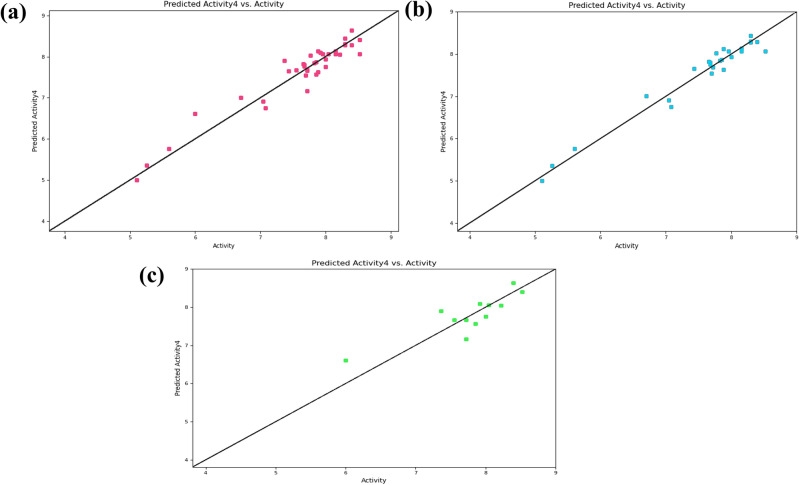
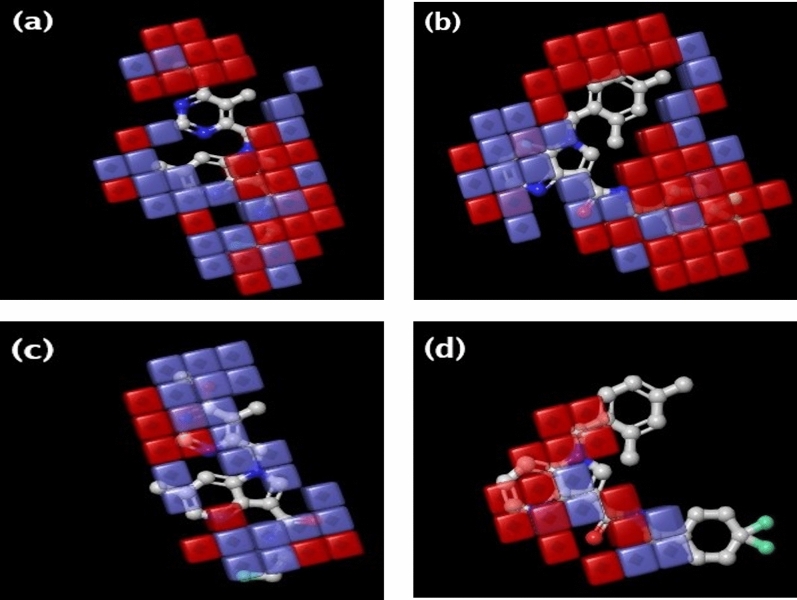
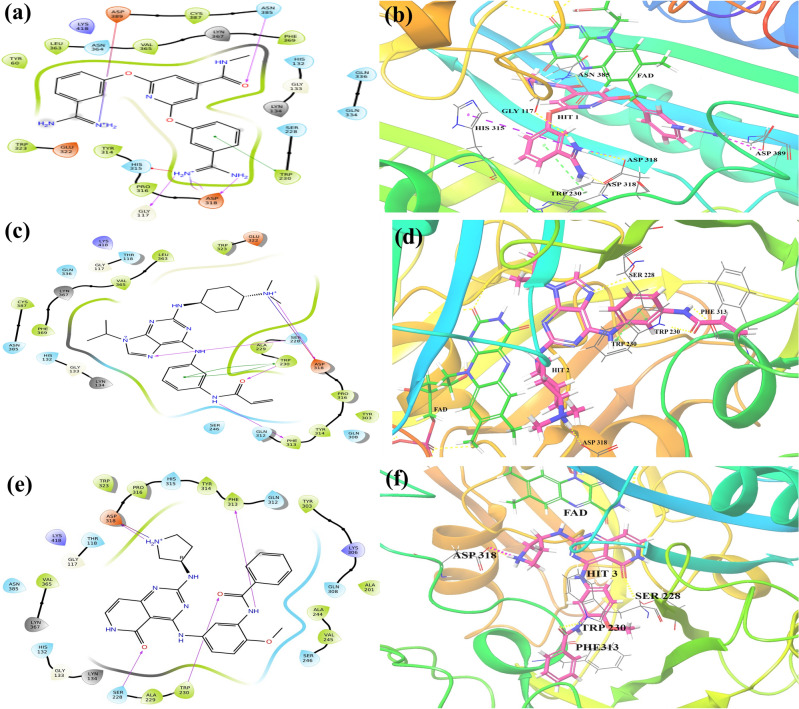
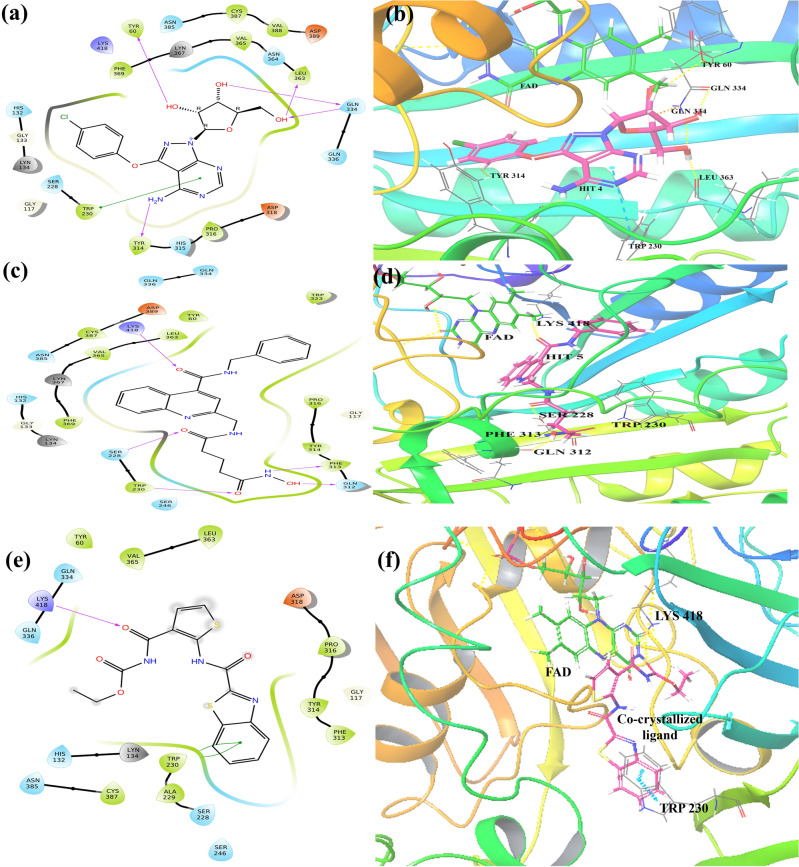
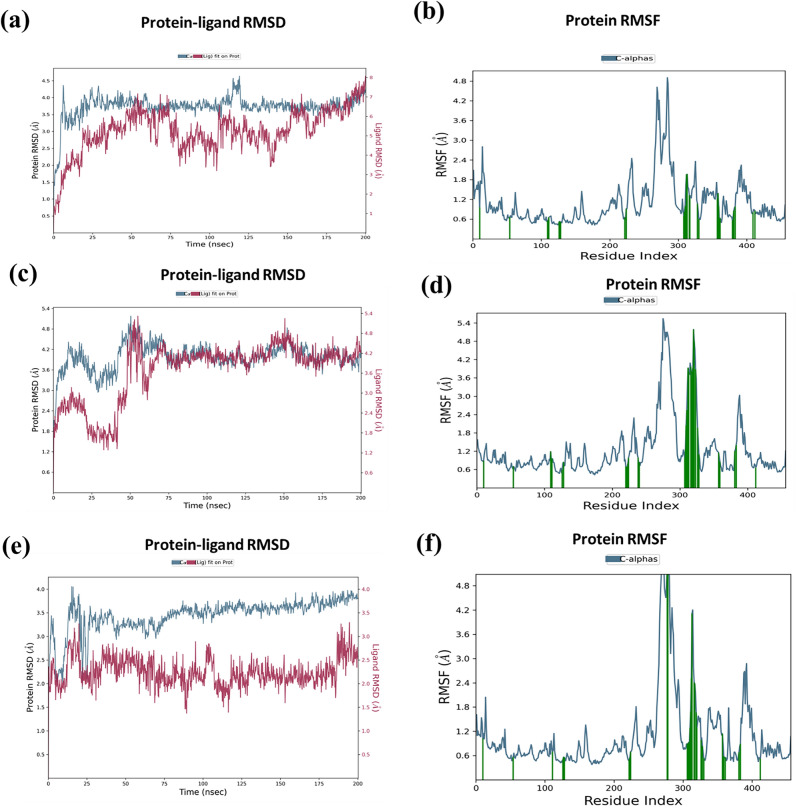
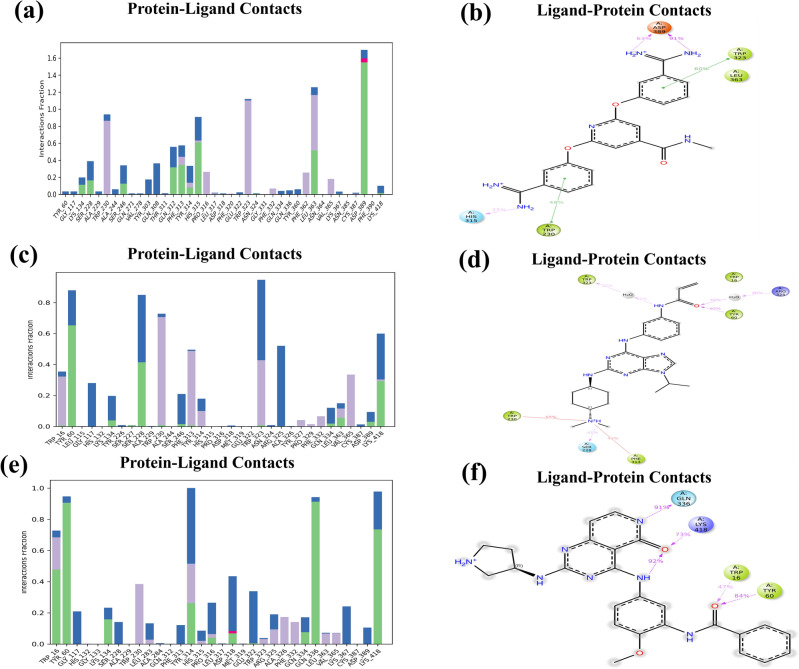
Decaprenylphosphoryl-β-D-ribose-2'-epimerase (DprE1), a crucial enzyme in the process of arabinogalactan and lipoarabinomannan biosynthesis, has become the target of choice for anti-TB drug discovery in the recent past. The current study aims to find the potential DprE1 inhibitors through in-silico approaches. Here, we built the pharmacophore and 3D-QSAR model using the reported 40 azaindole derivatives of DprE1 inhibitors. The best pharmacophore hypothesis (ADRRR_1) was employed for the virtual screening of the chEMBL database. To identify prospective hits, molecules with good phase scores (> 2.000) were further evaluated by molecular docking studies for their ability to bind to the DprE1 enzyme (PDB: 4KW5). Based on their binding affinities (< - 9.0 kcal/mole), the best hits were subjected to the calculation of free-binding energies (Prime/MM-GBSA), pharmacokinetic, and druglikeness evaluations. The top 10 hits retrieved from these results were selected to predict their inhibitory activities via the developed 3D-QSAR model with a regression coefficient (R) value of 0.9608 and predictive coefficient (Q) value of 0.7313. The induced fit docking (IFD) studies and in-silico prediction of anti-TB sensitivity for these top 10 hits were also implemented. Molecular dynamics simulations (MDS) were performed for the top 5 hit molecules for 200 ns to check the stability of the hits with DprE1. Based on their conformational stability throughout the 200 ns simulation, hit 2 (chEMBL_SDF:357100) was identified as the best hit against DprE1 with an accepted safety profile. The MD results were also in accordance with the docking score, MM-GBSA value, and 3D-QSAR predicted activity. The hit 2 molecule, (N-(3-((2-(((1r,4r)-4-(dimethylamino)cyclohexyl)amino)-9-isopropyl-9H-purin-6-yl)amino)phenyl)acrylamide) could serve as a lead for the discovery of a novel DprE1 inhibiting anti-TB drug.
去甲二磷酸-β-D-核糖-2'-差向异构酶(DprE1)是阿拉伯半乳聚糖和脂阿拉伯甘露聚糖生物合成过程中的关键酶,已成为最近抗结核药物发现的首选靶标。本研究旨在通过计算方法寻找潜在的 DprE1 抑制剂。在这里,我们使用报道的 40 种 DprE1 抑制剂的氮杂吲哚衍生物构建了药效团和 3D-QSAR 模型。最佳药效团假设(ADRRR_1)用于虚拟筛选 chEMBL 数据库。为了鉴定潜在的命中化合物,对具有良好相位分数(>2.000)的分子进行分子对接研究,以评估它们与 DprE1 酶(PDB:4KW5)结合的能力。基于它们的结合亲和力(< -9.0 kcal/mole),最佳命中化合物通过 Prime/MM-GBSA 计算自由结合能、药代动力学和类药性评估。从这些结果中检索到的前 10 个命中化合物被选为通过开发的 3D-QSAR 模型预测其抑制活性,该模型的回归系数(R)值为 0.9608,预测系数(Q)值为 0.7313。还对这 10 个最佳命中化合物进行了诱导契合对接(IFD)研究和抗结核敏感性的计算机预测。对前 5 个命中化合物进行了 200 ns 的分子动力学模拟(MDS),以检查它们与 DprE1 的结合稳定性。根据它们在整个 200 ns 模拟过程中的构象稳定性,命中化合物 2(chEMBL_SDF:357100)被确定为与 DprE1 结合的最佳命中化合物,具有可接受的安全性。MD 结果也与对接评分、MM-GBSA 值和 3D-QSAR 预测的活性一致。命中化合物 2 分子((N-(3-((2-(((1r,4r)-4-(二甲基氨基)环己基)氨基)-9-异丙基-9H-嘌呤-6-基)氨基)苯基)丙烯酰胺)可以作为发现新型 DprE1 抑制抗结核药物的先导化合物。