Center for Computational and Quantitative Genetics, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, 30322, USA.
Department of Biostatistics and Data Science, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX, 77030, USA.
Alzheimers Res Ther. 2024 Jun 1;16(1):120. doi: 10.1186/s13195-024-01488-7.
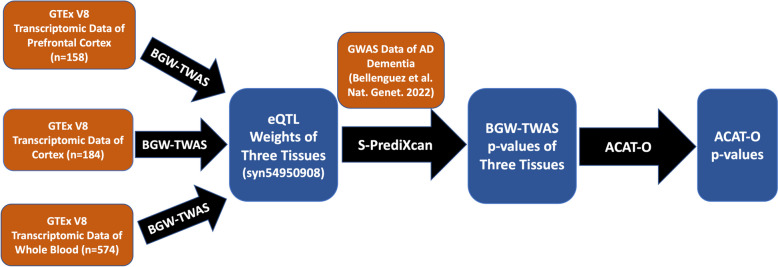
Transcriptome-wide association study (TWAS) is an influential tool for identifying genes associated with complex diseases whose genetic effects are likely mediated through transcriptome. TWAS utilizes reference genetic and transcriptomic data to estimate effect sizes of genetic variants on gene expression (i.e., effect sizes of a broad sense of expression quantitative trait loci, eQTL). These estimated effect sizes are employed as variant weights in gene-based association tests, facilitating the mapping of risk genes with genome-wide association study (GWAS) data. However, most existing TWAS of Alzheimer's disease (AD) dementia are limited to studying only cis-eQTL proximal to the test gene. To overcome this limitation, we applied the Bayesian Genome-wide TWAS (BGW-TWAS) method to leveraging both cis- and trans- eQTL of brain and blood tissues, in order to enhance mapping risk genes for AD dementia.
We first applied BGW-TWAS to the Genotype-Tissue Expression (GTEx) V8 dataset to estimate cis- and trans- eQTL effect sizes of the prefrontal cortex, cortex, and whole blood tissues. Estimated eQTL effect sizes were integrated with the summary data of the most recent GWAS of AD dementia to obtain BGW-TWAS (i.e., gene-based association test) p-values of AD dementia per gene per tissue type. Then we used the aggregated Cauchy association test to combine TWAS p-values across three tissues to obtain omnibus TWAS p-values per gene.
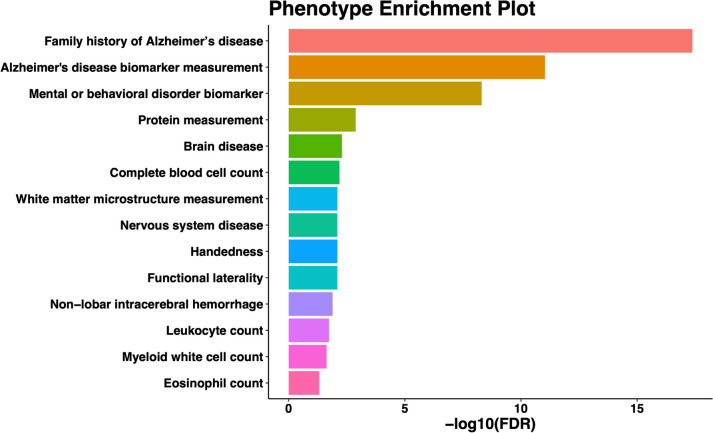
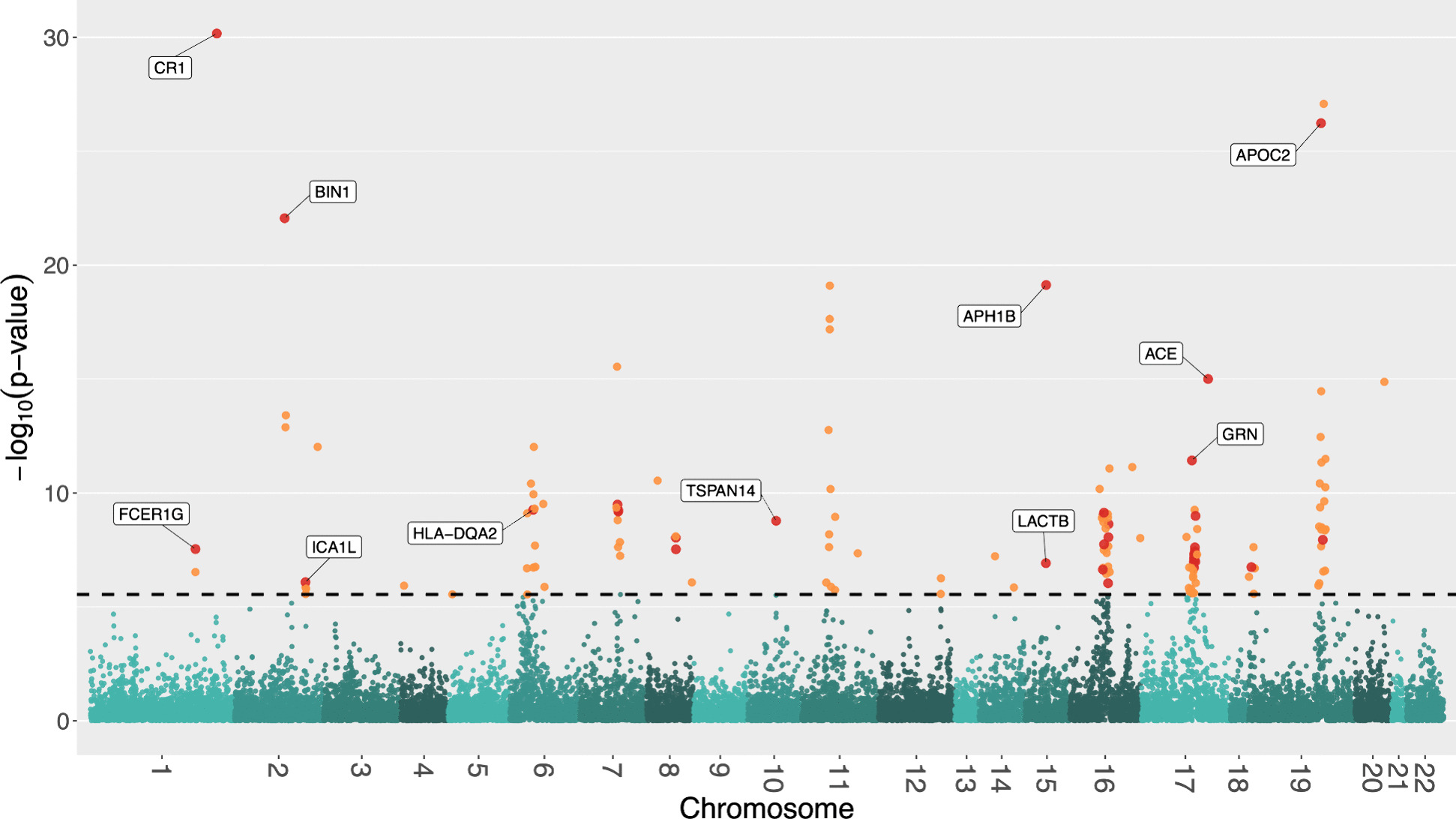
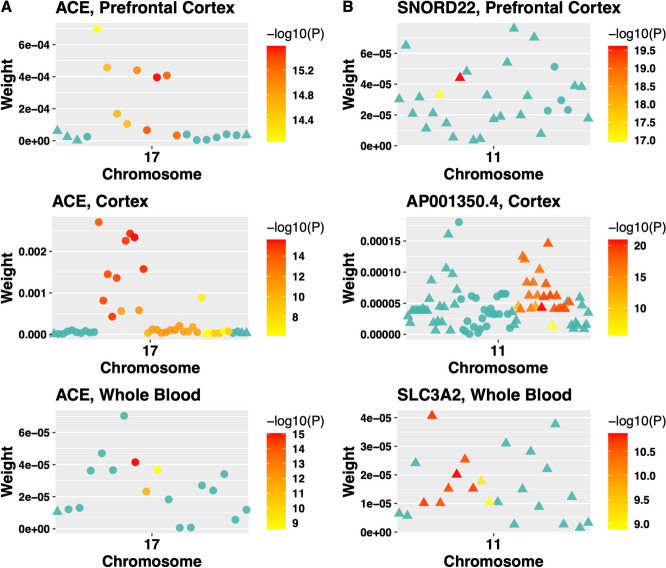
We identified 85 significant genes in prefrontal cortex, 82 in cortex, and 76 in whole blood that were significantly associated with AD dementia. By combining BGW-TWAS p-values across these three tissues, we obtained 141 significant risk genes including 34 genes primarily due to trans-eQTL and 35 mapped risk genes in GWAS Catalog. With these 141 significant risk genes, we detected functional clusters comprised of both known mapped GWAS risk genes of AD in GWAS Catalog and our identified TWAS risk genes by protein-protein interaction network analysis, as well as several enriched phenotypes related to AD.
We applied BGW-TWAS and aggregated Cauchy test methods to integrate both cis- and trans- eQTL data of brain and blood tissues with GWAS summary data, identifying 141 TWAS risk genes of AD dementia. These identified risk genes provide novel insights into the underlying biological mechanisms of AD dementia and potential gene targets for therapeutics development.
转录组全关联研究(TWAS)是一种有影响力的工具,可用于识别与复杂疾病相关的基因,这些基因的遗传效应可能通过转录组介导。TWAS 利用参考遗传和转录组数据来估计遗传变异对基因表达的效应大小(即广义表达数量性状基因座的效应大小,eQTL)。这些估计的效应大小被用作基于基因的关联测试中的变异权重,有助于利用全基因组关联研究(GWAS)数据对风险基因进行映射。然而,大多数现有的阿尔茨海默病(AD)痴呆 TWAS 仅限于研究仅位于测试基因附近的顺式-eQTL。为了克服这一限制,我们应用了贝叶斯全基因组 TWAS(BGW-TWAS)方法来利用大脑和血液组织中的顺式和反式-eQTL,以增强 AD 痴呆风险基因的映射。
我们首先将 BGW-TWAS 应用于基因型组织表达(GTEx)V8 数据集,以估计前额叶皮质、皮质和全血组织的顺式和反式-eQTL 效应大小。估计的 eQTL 效应大小与最近的 AD 痴呆 GWAS 的汇总数据相结合,以获得每个组织类型每个基因的 BGW-TWAS(即基于基因的关联测试)p 值。然后,我们使用聚合的 Cauchy 关联测试来合并三个组织的 TWAS p 值,以获得每个基因的总体 TWAS p 值。
我们在前额皮质中鉴定出 85 个与 AD 痴呆显著相关的基因,在皮质中鉴定出 82 个,在全血中鉴定出 76 个。通过合并这三个组织的 BGW-TWAS p 值,我们获得了 141 个显著的风险基因,其中包括 34 个主要由于反式-eQTL 引起的基因和 35 个在 GWAS 目录中映射的风险基因。利用这 141 个显著的风险基因,我们通过蛋白质-蛋白质相互作用网络分析检测到了包含已知映射的 AD GWAS 风险基因和我们通过 TWAS 识别的风险基因的功能簇,以及与 AD 相关的几个丰富表型。
我们应用了 BGW-TWAS 和聚合的 Cauchy 测试方法,将大脑和血液组织的顺式和反式-eQTL 数据与 GWAS 汇总数据相结合,确定了 141 个 AD 痴呆的 TWAS 风险基因。这些鉴定出的风险基因为 AD 痴呆的潜在生物学机制提供了新的见解,并为治疗药物的开发提供了潜在的基因靶点。