Cantin Lindsey J, Gregory Vanessa, Blum Laura N, Foster Jeremy M
Biochemistry and Microbiology Division, New England BioLabs, Ipswich, MA, United States.
Applications and Product Development, New England BioLabs, Ipswich, MA, United States.
Front Microbiol. 2024 May 20;15:1418032. doi: 10.3389/fmicb.2024.1418032. eCollection 2024.
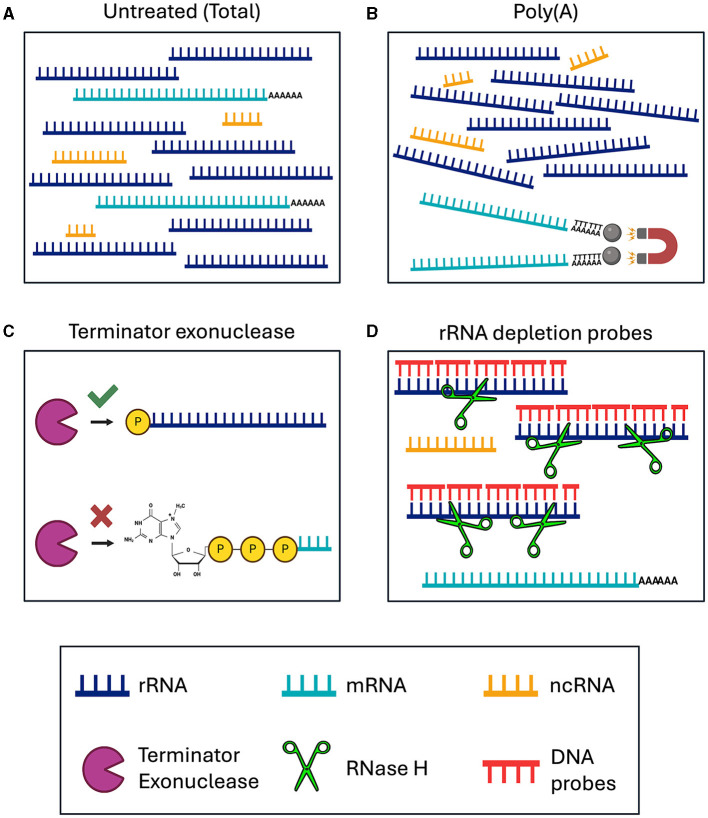
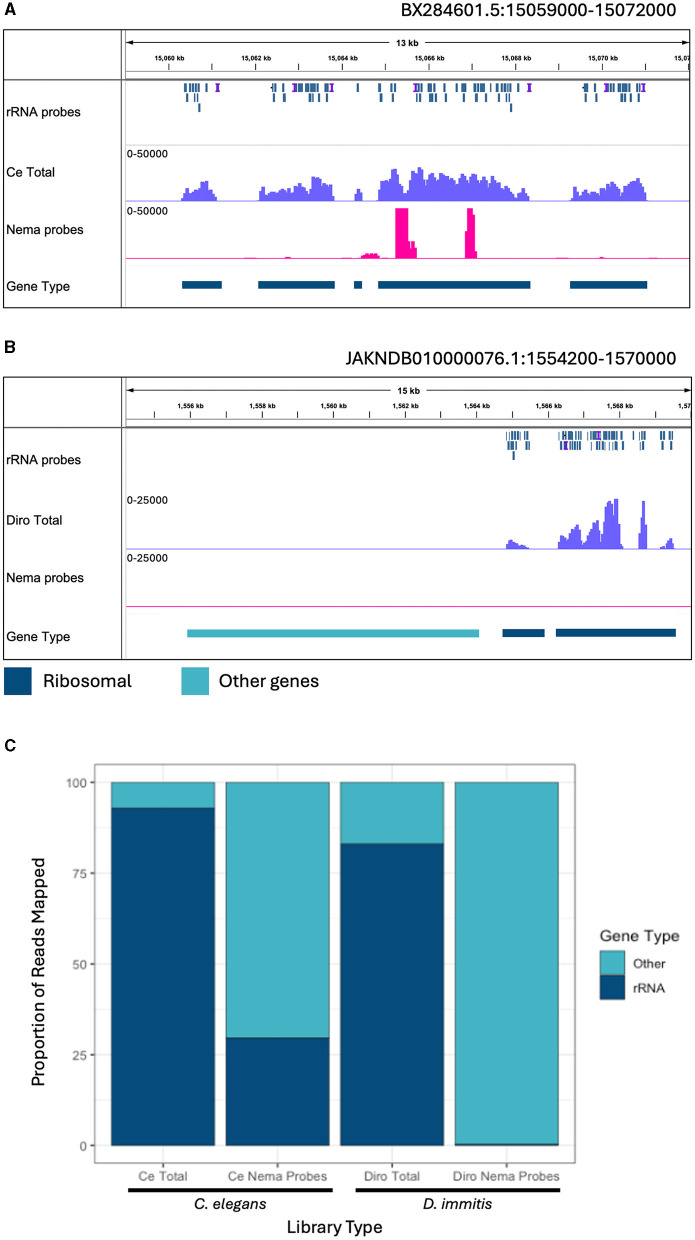
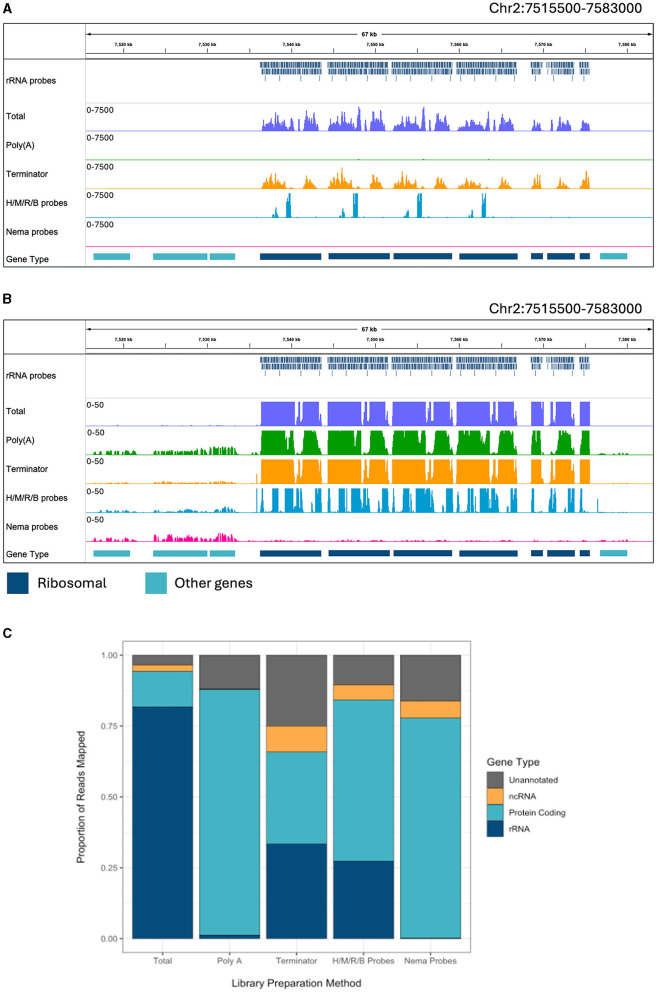
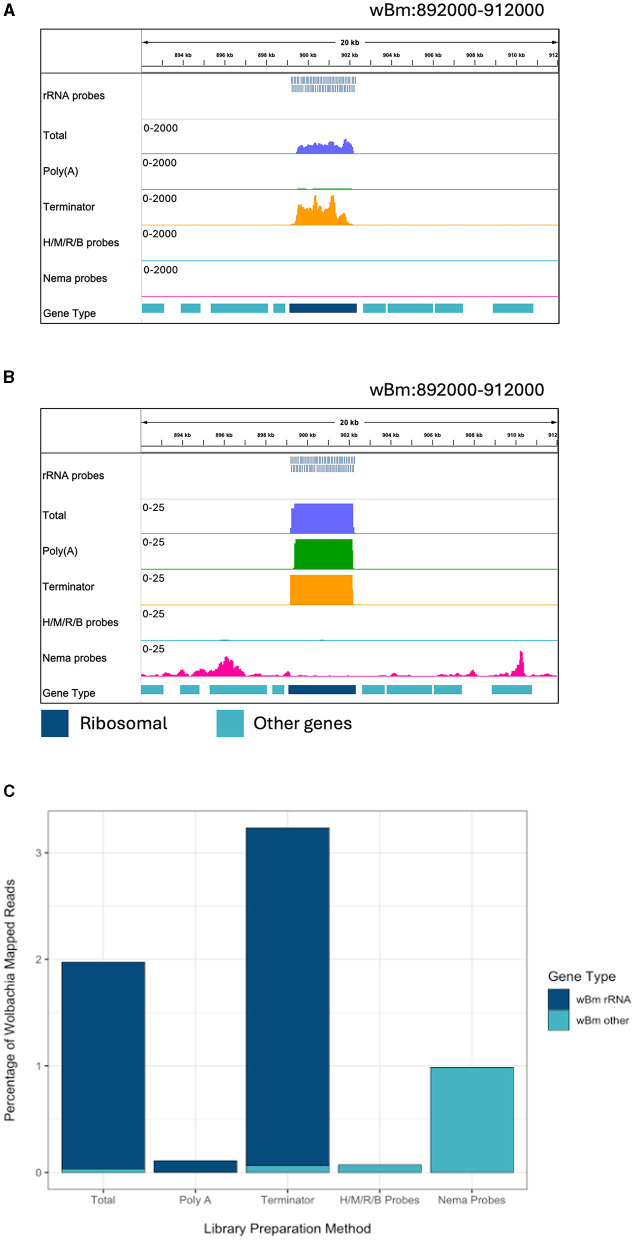
Lymphatic filariasis is caused by parasitic nematodes and is a leading cause of disability worldwide. Many filarial worms contain the bacterium as an obligate endosymbiont. RNA sequencing is a common technique used to study their molecular relationships and to identify potential drug targets against the nematode and bacteria. Ribosomal RNA (rRNA) is the most abundant RNA species, accounting for 80-90% of the RNA in a sample. To reduce sequencing costs, it is necessary to remove ribosomal reads through poly-A enrichment or ribosomal depletion. Bacterial RNA does not contain a poly-A tail, making it difficult to sequence both the nematode and from the same library preparation using standard poly-A selection. Ribosomal depletion can utilize species-specific oligonucleotide probes to remove rRNA through pull-down or degradation methods. While species-specific probes are commercially available for many commonly studied model organisms, there are currently limited depletion options for filarial parasites. Here, we performed total RNA sequencing from containing the symbiont (Bm) and designed ssDNA depletion probes against their rRNA sequences. We compared the total RNA library to poly-A enriched, Terminator 5'-Phosphate-Dependent Exonuclease treated, NEBNext Human/Bacteria rRNA depleted and our custom nematode probe depleted libraries. The custom nematode depletion library had the lowest percentage of ribosomal reads across all methods, with a 300-fold decrease in rRNA when compared to the total RNA library. The nematode depletion libraries also contained the highest percentage of mRNA reads, resulting in a 16-1,000-fold increase in bacterial reads compared to the other enrichment and depletion methods. Finally, we found that the depletion probes can remove rRNA from the filarial worm and the majority of rRNA from the more distantly related free living nematode . These custom filarial probes will allow for future dual RNA-seq experiments between nematodes and their bacterial symbionts from a single sequencing library.
淋巴丝虫病由寄生线虫引起,是全球导致残疾的主要原因。许多丝虫含有一种细菌作为专性内共生菌。RNA测序是一种常用技术,用于研究它们的分子关系,并识别针对线虫和细菌的潜在药物靶点。核糖体RNA(rRNA)是最丰富的RNA种类,占样本中RNA的80 - 90%。为降低测序成本,有必要通过多聚腺苷酸富集或核糖体去除来去除核糖体读数。细菌RNA不包含多聚腺苷酸尾,这使得使用标准的多聚腺苷酸选择方法从同一文库制备中对线虫和细菌进行测序变得困难。核糖体去除可以利用物种特异性寡核苷酸探针通过下拉或降解方法去除rRNA。虽然针对许多常用研究模式生物有商业化的物种特异性探针,但目前针对丝虫寄生虫的去除选项有限。在这里,我们对含有共生菌(Bm)的样本进行了总RNA测序,并针对其rRNA序列设计了单链DNA去除探针。我们将总RNA文库与多聚腺苷酸富集文库、依赖5'-磷酸的外切核酸酶处理文库、NEBNext人类/细菌rRNA去除文库以及我们定制的线虫探针去除文库进行了比较。在所有方法中,定制的线虫去除文库的核糖体读数百分比最低,与总RNA文库相比,rRNA减少了300倍。线虫去除文库中细菌mRNA读数的百分比也最高,与其他富集和去除方法相比,细菌读数增加了16 - 1000倍。最后,我们发现这些去除探针可以从丝虫中去除rRNA,并从亲缘关系更远的自由生活线虫中去除大部分rRNA。这些定制的丝虫探针将有助于未来从单个测序文库对线虫及其细菌共生体进行双RNA测序实验。