Suhre Karsten, Chen Qingwen, Halama Anna, Mendez Kevin, Dahlin Amber, Stephan Nisha, Thareja Gaurav, Sarwath Hina, Guturu Harendra, Dwaraka Varun B, Batzoglou Serafim, Schmidt Frank, Lasky-Su Jessica A
Bioinformatics Core, Weill Cornell Medicine-Qatar, Education City, 24144 Doha, Qatar.
Englander Institute for Precision Medicine, Weill Cornell Medicine, New York, NY 10021, USA.
bioRxiv. 2024 Jun 1:2024.05.27.596028. doi: 10.1101/2024.05.27.596028.
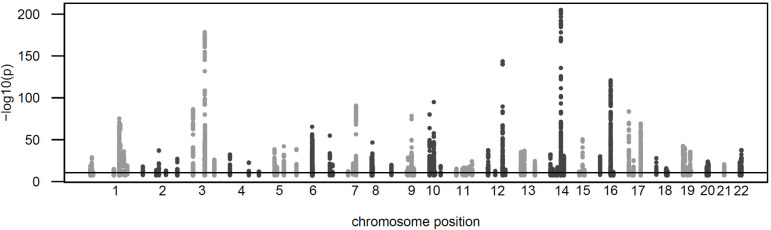
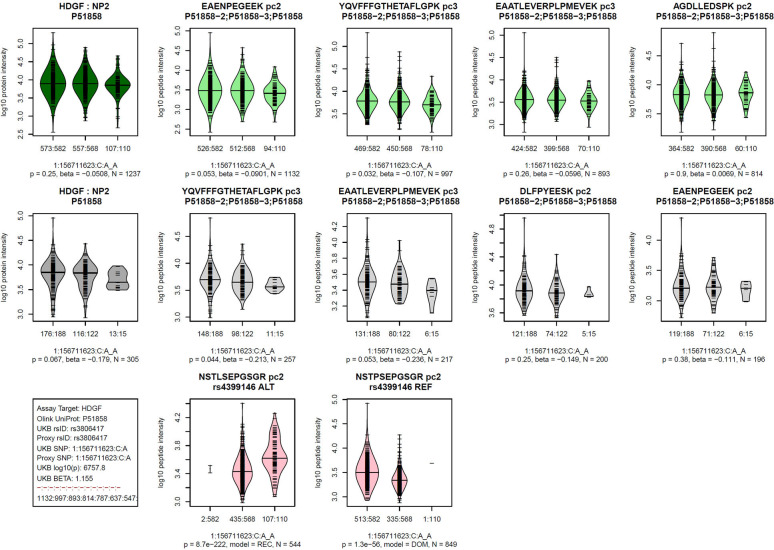
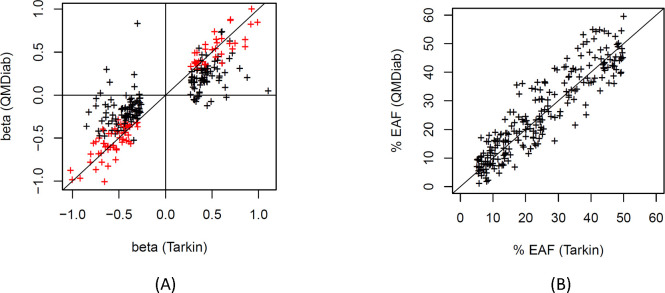
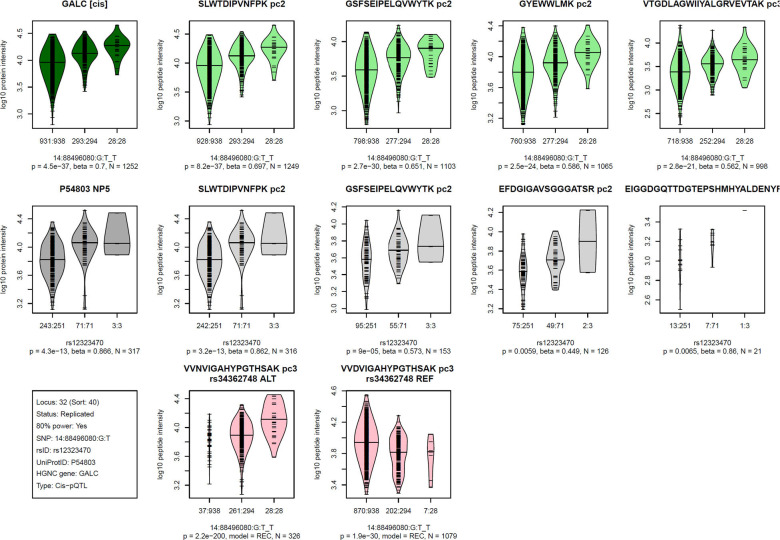
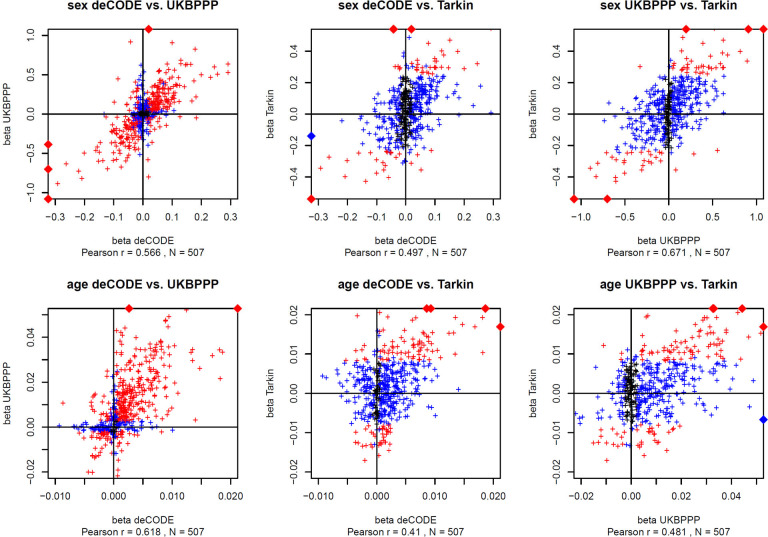
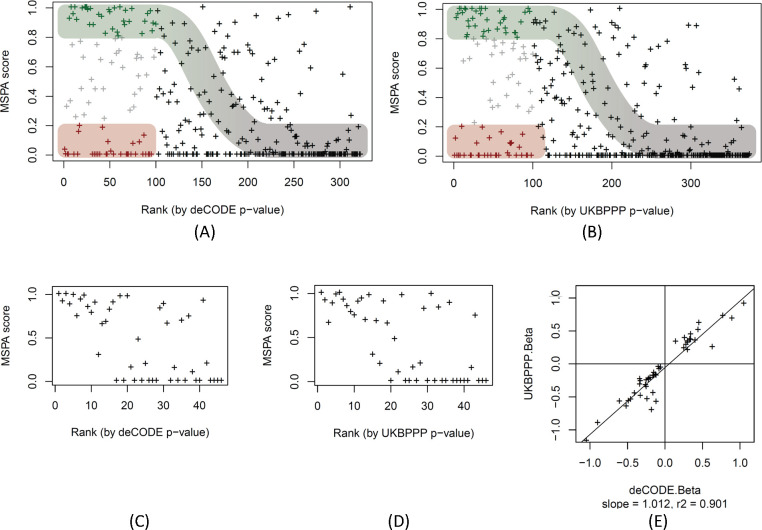
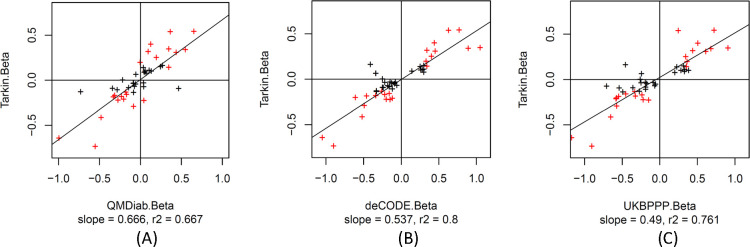
Genome-wide association studies (GWAS) with proteomics are essential tools for drug discovery. To date, most studies have used affinity proteomics platforms, which have limited discovery to protein panels covered by the available affinity binders. Furthermore, it is not clear to which extent protein epitope changing variants interfere with the detection of protein quantitative trait loci (pQTLs). Mass spectrometry-based (MS) proteomics can overcome some of these limitations. Here we report a GWAS using the MS-based Seer Proteograph platform with blood samples from a discovery cohort of 1,260 American participants and a replication in 325 individuals from Asia, with diverse ethnic backgrounds. We analysed 1,980 proteins quantified in at least 80% of the samples, out of 5,753 proteins quantified across the discovery cohort. We identified 252 and replicated 90 pQTLs, where 30 of the replicated pQTLs have not been reported before. We further investigated 200 of the strongest associated cis-pQTLs previously identified using the SOMAscan and the Olink platforms and found that up to one third of the affinity proteomics pQTLs may be affected by epitope effects, while another third were confirmed by MS proteomics to be consistent with the hypothesis that genetic variants induce changes in protein expression. The present study demonstrates the complementarity of the different proteomics approaches and reports pQTLs not accessible to affinity proteomics, suggesting that many more pQTLs remain to be discovered using MS-based platforms.
全基因组关联研究(GWAS)与蛋白质组学相结合是药物发现的重要工具。迄今为止,大多数研究使用的是亲和蛋白质组学平台,其发现范围仅限于现有亲和结合剂所覆盖的蛋白质组。此外,尚不清楚蛋白质表位变化变体在多大程度上干扰蛋白质定量性状位点(pQTL)的检测。基于质谱(MS)的蛋白质组学可以克服其中一些局限性。在此,我们报告了一项GWAS,使用基于MS的Seer Proteograph平台,对来自1260名美国参与者的发现队列的血液样本进行分析,并在325名来自亚洲、具有不同种族背景的个体中进行复制。我们分析了在发现队列中定量的5753种蛋白质中,至少在80%的样本中定量的1980种蛋白质。我们鉴定出252个pQTL并复制了90个,其中30个复制的pQTL此前未被报道过。我们进一步研究了之前使用SOMAscan和Olink平台鉴定出的200个最强相关的顺式pQTL,发现高达三分之一的亲和蛋白质组学pQTL可能受表位效应影响,而另外三分之一经MS蛋白质组学证实与遗传变异诱导蛋白质表达变化的假设一致。本研究证明了不同蛋白质组学方法的互补性,并报告了亲和蛋白质组学无法获得的pQTL,这表明使用基于MS的平台可能还有更多pQTL有待发现。