Department of Pharmacology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Department of Internal Medicine (Nephrology), University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Mol Metab. 2024 Aug;86:101967. doi: 10.1016/j.molmet.2024.101967. Epub 2024 Jun 12.
In response to bacterial inflammation, anorexia of acute illness is protective and is associated with the induction of fasting metabolic programs such as ketogenesis. Forced feeding during the anorectic period induced by bacterial inflammation is associated with suppressed ketogenesis and increased mortality. As ketogenesis is considered essential in fasting adaptation, we sought to determine the role of ketogenesis in illness-induced anorexia.
A mouse model of inducible hepatic specific deletion of the rate limiting enzyme for ketogenesis (HMG-CoA synthase 2, Hmgcs2) was used to investigate the role of ketogenesis in endotoxemia, a model of bacterial inflammation, and in prolonged starvation.
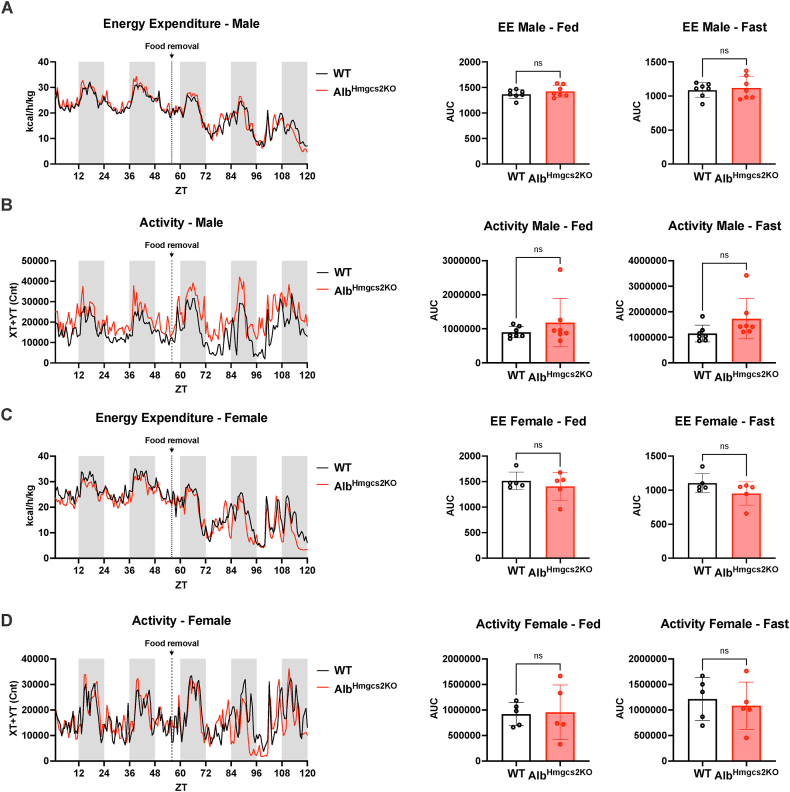
Mice deficient of hepatic Hmgcs2 failed to develop ketosis during endotoxemia and during prolonged fasting. Surprisingly, hepatic HMGCS2 deficiency and the lack of ketosis did not affect survival, glycemia, or body temperature in response to endotoxemia. Mice with hepatic ketogenic deficiency also did not exhibit any defects in starvation adaptation and were able to maintain blood glucose, body temperature, and lean mass compared to littermate wild-type controls. Mice with hepatic HMGCS2 deficiency exhibited higher levels of plasma acetate levels in response to fasting.
Circulating hepatic-derived ketones do not provide protection against endotoxemia, suggesting that alternative mechanisms drive the increased mortality from forced feeding during illness-induced anorexia. Hepatic ketones are also dispensable for surviving prolonged starvation in the absence of inflammation. Our study challenges the notion that hepatic ketogenesis is required to maintain blood glucose and preserve lean mass during starvation, raising the possibility of extrahepatic ketogenesis and use of alternative fuels as potential means of metabolic compensation.
针对细菌炎症,急性疾病的厌食是一种保护机制,与禁食代谢程序如酮体生成的诱导有关。在细菌炎症引起的厌食期进行强制喂食与酮体生成受抑制和死亡率增加有关。由于酮体生成被认为是禁食适应所必需的,我们试图确定酮体生成在疾病引起的厌食中的作用。
使用诱导性肝特异性缺失酮体生成限速酶(羟甲戊二酰辅酶 A 合酶 2,Hmgcs2)的小鼠模型,研究酮体生成在内毒素血症(细菌炎症模型)和长期饥饿中的作用。
缺乏肝 Hmgcs2 的小鼠在内毒素血症和长期禁食期间无法产生酮症。令人惊讶的是,肝 HMGCS2 缺乏和缺乏酮症并不影响对内毒素血症的存活、血糖或体温。肝酮生成缺乏的小鼠在饥饿适应方面也没有任何缺陷,并且能够维持血糖、体温和瘦体重,与同窝野生型对照相比。与野生型对照相比,缺乏肝 HMGCS2 的小鼠在禁食时表现出更高的血浆乙酸盐水平。
循环肝源性酮体不能提供对内毒素血症的保护,这表明在疾病引起的厌食期间进行强制喂食导致的死亡率增加可能有替代机制。在没有炎症的情况下,肝酮体对于长时间的饥饿生存也是可有可无的。我们的研究挑战了肝酮体生成对于维持饥饿期间血糖和保留瘦体重是必需的观点,提出了额外的肝外酮体生成和使用替代燃料作为潜在代谢补偿的可能性。