Department of Pharmacology, New York Medical College, Valhalla, New York, USA.
Department of Pharmacology and Toxicology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, State University of New York, Buffalo, New York, USA.
J Biol Chem. 2024 Jul;300(7):107460. doi: 10.1016/j.jbc.2024.107460. Epub 2024 Jun 12.
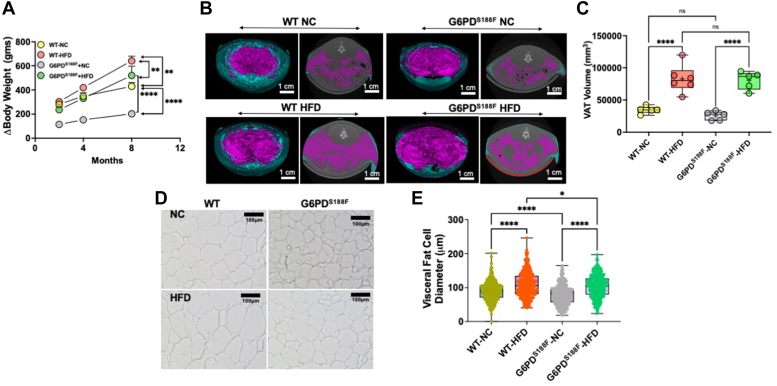
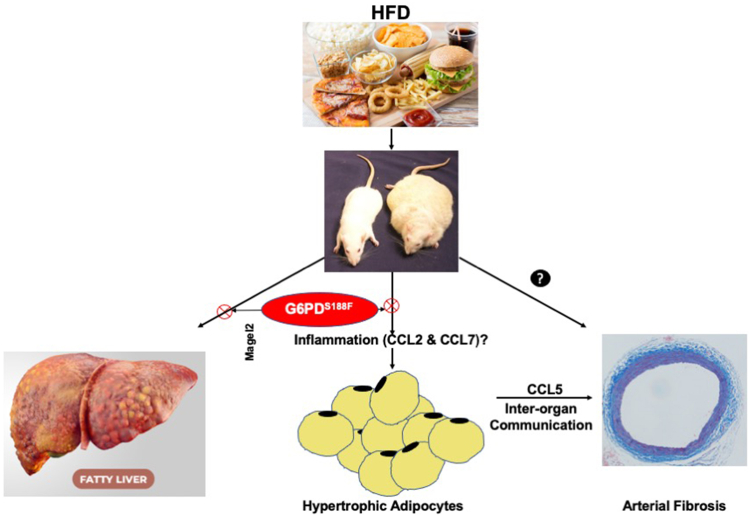
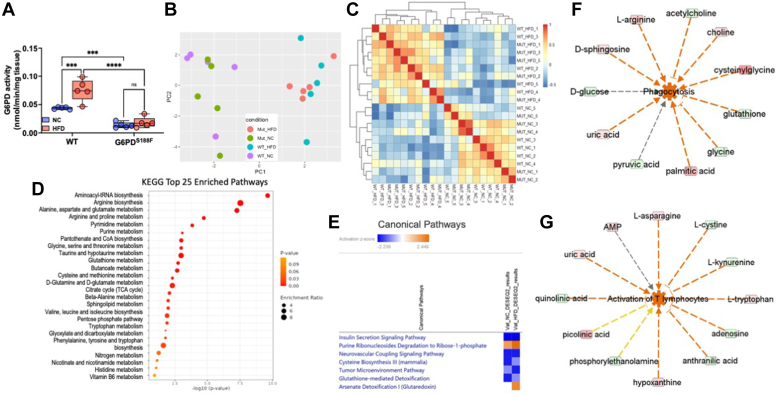
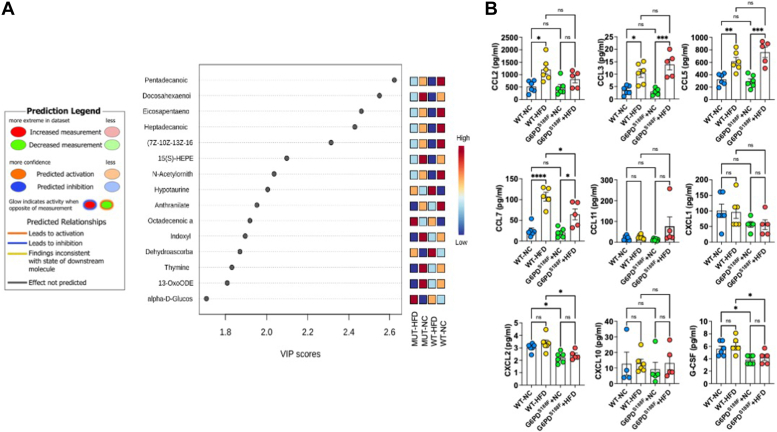
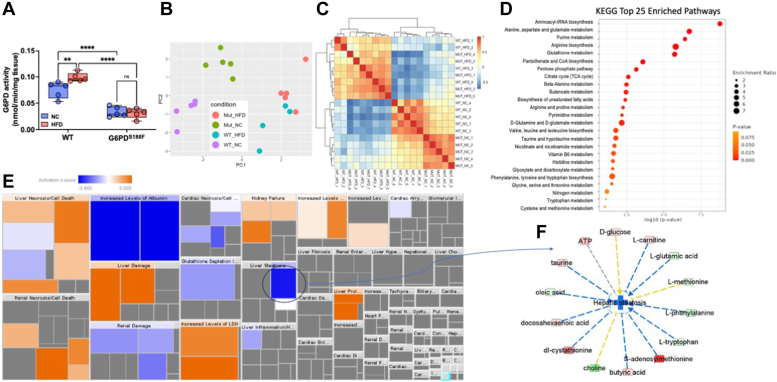
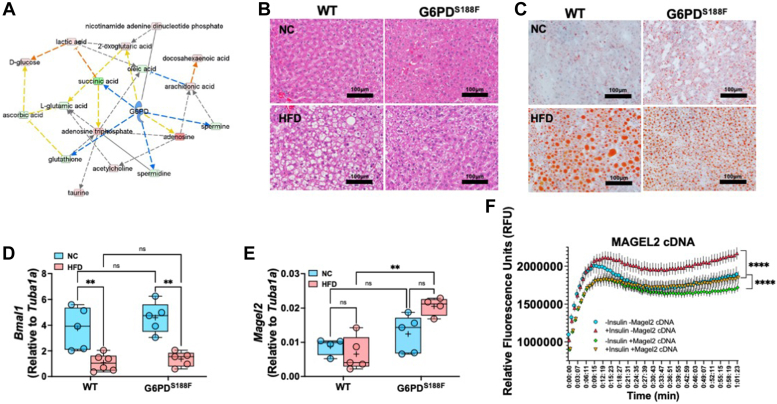
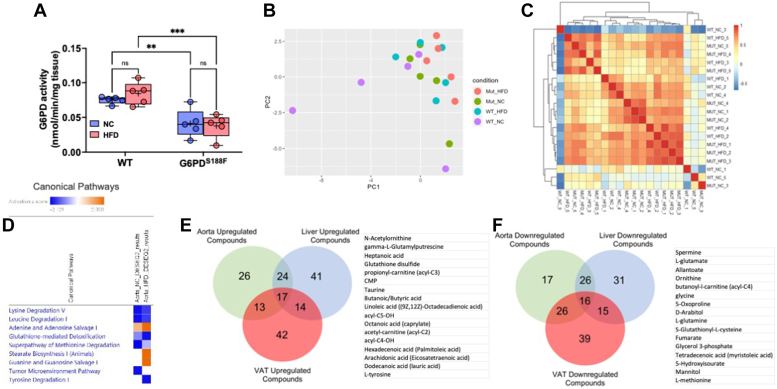
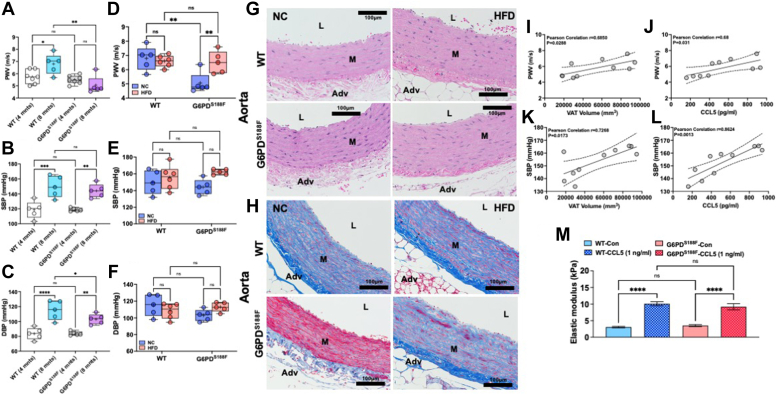
Obesity is a major risk factor for liver and cardiovascular diseases. However, obesity-driven mechanisms that contribute to the pathogenesis of multiple organ diseases are still obscure and treatment is inadequate. We hypothesized that increased , glucose-6-phosphate dehydrogenase (G6PD), the key rate-limiting enzyme in the pentose shunt, is critical in evoking metabolic reprogramming in multiple organs and is a significant contributor to the pathogenesis of liver and cardiovascular diseases. G6PD is induced by a carbohydrate-rich diet and insulin. Long-term (8 months) high-fat diet (HFD) feeding increased body weight and elicited metabolic reprogramming in visceral fat, liver, and aorta, of the wild-type rats. In addition, HFD increased inflammatory chemokines in visceral fat. Interestingly, CRISPR-edited loss-of-function Mediterranean G6PD variant (G6PD) rats, which mimic human polymorphism, moderated HFD-induced weight gain and metabolic reprogramming in visceral fat, liver, and aorta. The G6PD variant prevented HFD-induced CCL7 and adipocyte hypertrophy. Furthermore, the G6PD variant increased Magel2 - a gene encoding circadian clock-related protein that suppresses obesity associated with Prader-Willi syndrome - and reduced HFD-induced non-alcoholic fatty liver. Additionally, the G6PD variant reduced aging-induced aortic stiffening. Our findings suggest G6PD is a regulator of HFD-induced obesity, adipocyte hypertrophy, and fatty liver.
肥胖是肝脏和心血管疾病的主要危险因素。然而,导致多种器官疾病发病机制的肥胖驱动机制仍不清楚,治疗也不充分。我们假设增加葡萄糖-6-磷酸脱氢酶(G6PD),戊糖磷酸途径的关键限速酶,在多个器官中引发代谢重编程是至关重要的,并且是肝脏和心血管疾病发病机制的重要贡献者。G6PD 由富含碳水化合物的饮食和胰岛素诱导。长期(8 个月)高脂肪饮食(HFD)喂养增加了野生型大鼠的体重,并引发了内脏脂肪、肝脏和主动脉的代谢重编程。此外,HFD 增加了内脏脂肪中的炎症趋化因子。有趣的是,CRISPR 编辑的丧失功能的地中海 G6PD 变体(G6PD)大鼠模拟了人类多态性,适度了 HFD 诱导的体重增加和内脏脂肪、肝脏和主动脉的代谢重编程。G6PD 变体阻止了 HFD 诱导的 CCL7 和脂肪细胞肥大。此外,G6PD 变体增加了 Magel2——一种编码与生物钟相关的蛋白质的基因,该蛋白质抑制与普拉德-威利综合征相关的肥胖——并减少了 HFD 诱导的非酒精性脂肪肝。此外,G6PD 变体减少了衰老引起的主动脉僵硬。我们的研究结果表明,G6PD 是 HFD 诱导肥胖、脂肪细胞肥大和脂肪肝的调节剂。