Baumann Alexandra, Ruckert Christian, Meier Christoph, Hutschenreiter Tim, Remy Robert, Schnur Benedikt, Döbel Marvin, Fankep Rudel Christian Nkouamedjo, Skowronek Dariush, Kutz Oliver, Arnold Norbert, Katzke Anna-Lena, Forster Michael, Kobiela Anna-Lena, Thiedig Katharina, Zimmer Andreas, Ritter Julia, Weber Bernhard H F, Honisch Ellen, Hackmann Karl, Schmidt Gunnar, Sturm Marc, Ernst Corinna
Institute for Clinical Genetics, University Hospital Carl Gustav Carus at TUD Dresden University of Technology and Faculty of Medicine of TUD Dresden University of Technology, Dresden, Germany.
ERN GENTURIS, Hereditary Cancer Syndrome Center Dresden, Dresden, Germany.
Eur J Hum Genet. 2024 Aug;32(8):987-997. doi: 10.1038/s41431-024-01647-2. Epub 2024 Jun 21.
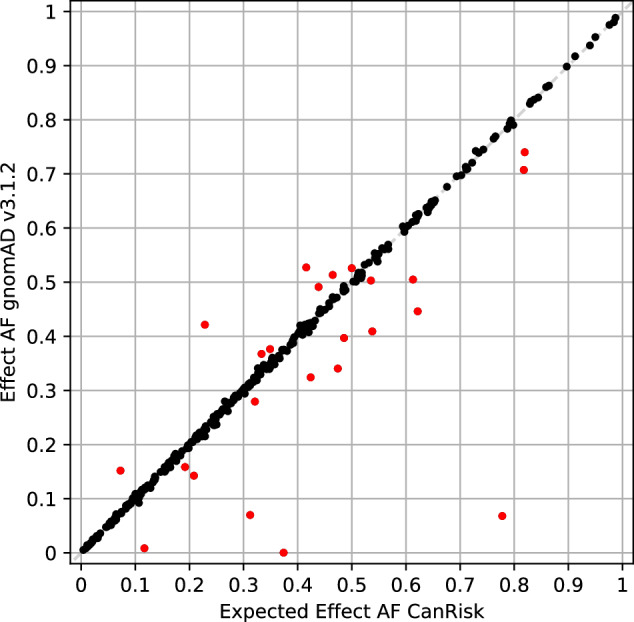
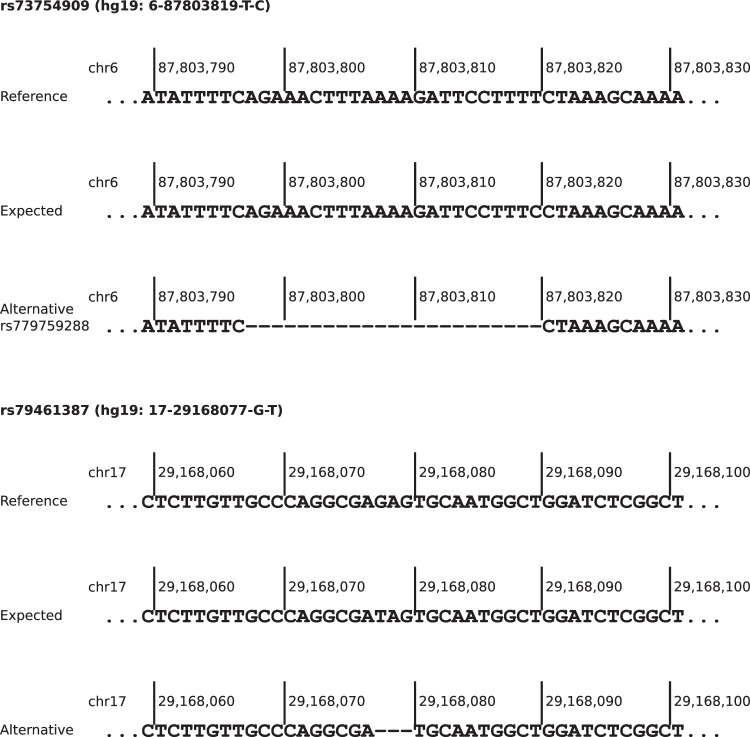
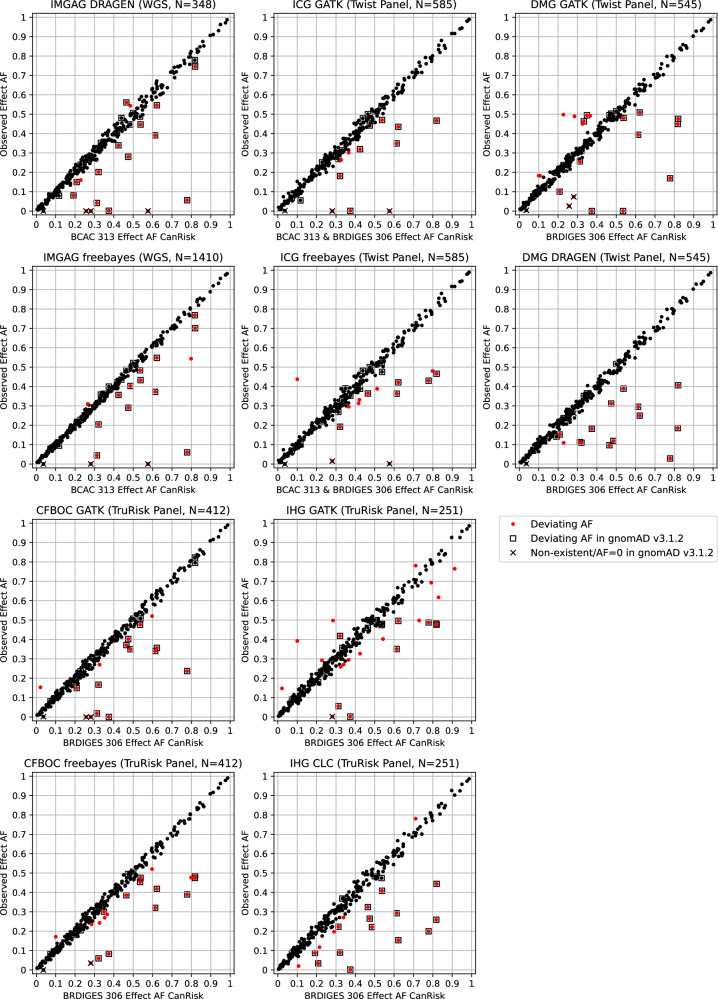
Considering polygenic risk scores (PRSs) in individual risk prediction is increasingly implemented in genetic testing for hereditary breast cancer (BC) based on next-generation sequencing (NGS). To calculate individual BC risks, the Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA) with the inclusion of the BCAC 313 or the BRIDGES 306 BC PRS is commonly used. The PRS calculation depends on accurately reproducing the variant allele frequencies (AFs) and, consequently, the distribution of PRS values anticipated by the algorithm. Here, the 324 loci of the BCAC 313 and the BRIDGES 306 BC PRS were examined in population-specific database gnomAD and in real-world data sets of five centers of the German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC), to determine whether these expected AFs can be reproduced by NGS-based genotyping. Four PRS loci were non-existent in gnomAD v3.1.2 non-Finnish Europeans, further 24 loci showed noticeably deviating AFs. In real-world data, between 11 and 23 loci were reported with noticeably deviating AFs, and were shown to have effects on final risk prediction. Deviations depended on the sequencing approach, variant caller and calling mode (forced versus unforced) employed. Therefore, this study demonstrates the necessity to apply quality assurance not only in terms of sequencing coverage but also observed AFs in a sufficiently large cohort, when implementing PRSs in a routine diagnostic setting. Furthermore, future PRS design should be guided by the technical reproducibility of expected AFs across commonly used genotyping methods, especially NGS, in addition to the observed effect sizes.
在遗传性乳腺癌(BC)的基因检测中,基于下一代测序(NGS)在个体风险预测中考虑多基因风险评分(PRSs)的情况越来越普遍。为了计算个体患BC的风险,通常使用包含BCAC 313或BRIDGES 306 BC PRS的乳腺癌和卵巢癌疾病发病率及携带者估计算法(BOADICEA)。PRS的计算依赖于准确再现变异等位基因频率(AFs),进而依赖于算法预期的PRS值分布。在此,研究人员在特定人群数据库gnomAD以及德国遗传性乳腺癌和卵巢癌联盟(GC-HBOC)五个中心的真实数据集里,对BCAC 313和BRIDGES 306 BC PRS的324个位点进行了检测,以确定基于NGS的基因分型能否再现这些预期的AFs。在gnomAD v3.1.2非芬兰欧洲人群中,有4个PRS位点不存在,另有24个位点显示出明显偏离的AFs。在真实数据中,有11至23个位点被报告AFs明显偏离,且显示对最终风险预测有影响。偏差取决于所采用的测序方法、变异检测软件和检测模式(强制与非强制)。因此,本研究表明,在常规诊断环境中实施PRS时,不仅要在测序覆盖度方面,而且要在足够大的队列中观察AFs,应用质量保证是必要的。此外,未来PRS的设计除了考虑观察到的效应大小外,还应以常用基因分型方法(尤其是NGS)中预期AFs的技术可重复性为指导。