Department of Neurology and Neuroscience Center, The First Affiliated Hospital of Jilin University, Changchun, China.
Front Immunol. 2024 Jul 3;15:1378130. doi: 10.3389/fimmu.2024.1378130. eCollection 2024.
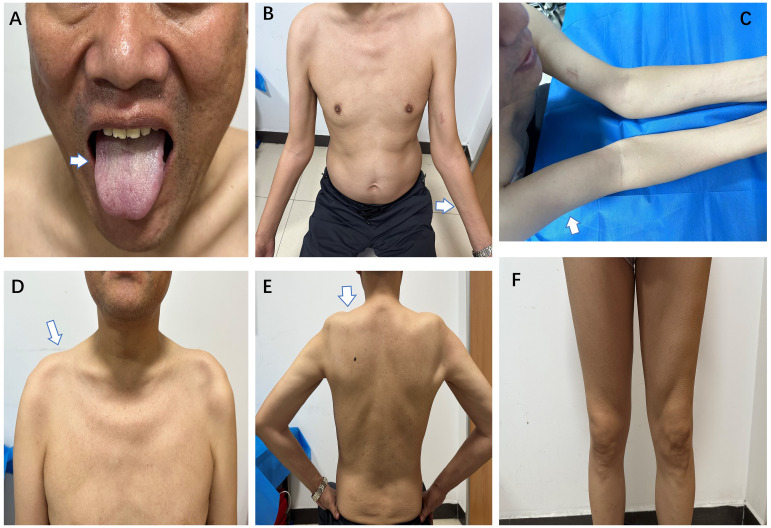
Brachio-cervical inflammatory myopathy (BCIM) is a rare inflammatory myopathy characterized by dysphagia, bilateral upper limb atrophy, limb-girdle muscle weakness, and myositis-specific antibody (MSA) negativity. BCIM has a low incidence and is commonly associated with autoimmune diseases. We present a case report of a 55-year-old man with progressive upper limb weakness and atrophy, diagnosed with flail arm syndrome (FAS). The initial electromyography revealed extensive spontaneous muscle activity and increased duration of motor unit potentials (MUPs). During follow-up, evidence of myogenic damage was observed, as indicated by a decreased duration of MUPs in the right biceps muscle. Laboratory and genetic testing ruled out hereditary or acquired diseases. Negative serological antibodies for myasthenia gravis. Hereditary or acquired diseases were ruled out through laboratory and genetic testing. Whole-body muscle magnetic resonance imaging (MRI) showed extensive edema and fat replacement in the bilateral upper limbs, scapular, and central axis muscles, while the lower extremities were relatively mildly affected. Muscle biopsy revealed numerous foci of inflammatory cells distributed throughout the muscle bundle, with predominant CD20, CD138, and CD68 expression, accompanied by a light infiltration of CD3 and CD4 expression. The muscle weakness improved with the combination of oral prednisone (initially 60 mg/day, tapered) and methotrexate (5 mg/week) treatment.
颈臂肌炎性肌病(BCIM)是一种罕见的炎性肌病,其特征为吞咽困难、双侧上肢萎缩、肢体带肌无力和肌炎特异性抗体(MSA)阴性。BCIM 的发病率较低,常与自身免疫性疾病相关。我们报告了一例 55 岁男性进行性上肢无力和萎缩的病例,诊断为连枷臂综合征(FAS)。初始肌电图显示广泛的自发性肌肉活动和运动单位电位(MUP)持续时间增加。在随访过程中,观察到肌源性损伤的证据,右侧肱二头肌 MUP 持续时间减少。实验室和基因检测排除了遗传性或获得性疾病。抗重症肌无力抗体阴性。通过实验室和基因检测排除了遗传性或获得性疾病。全身肌肉磁共振成像(MRI)显示双侧上肢、肩胛带和中轴肌肉广泛水肿和脂肪替代,下肢受累相对较轻。肌肉活检显示大量炎性细胞分布在整个肌束中,主要表达 CD20、CD138 和 CD68,伴有 CD3 和 CD4 表达的轻度浸润。肌肉无力在口服泼尼松(初始剂量 60mg/天,逐渐减少)和甲氨蝶呤(5mg/周)联合治疗后得到改善。