Yang Fan, Ling Shenglong, Zhou Yingxin, Zhang Yanan, Lv Pei, Liu Sanling, Fang Wei, Sun Wenjing, Hu Liaoyuan A, Zhang Longhua, Shi Pan, Tian Changlin
Hefei National Laboratory of Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei 230026, China.
Amgen Asia R&D Center, Amgen Research, Shanghai 201210, China.
Natl Sci Rev. 2020 Nov 24;8(9):nwaa284. doi: 10.1093/nsr/nwaa284. eCollection 2021 Sep.
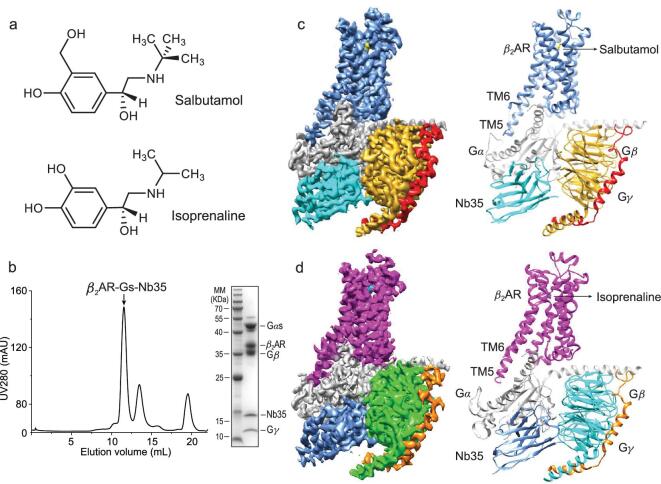
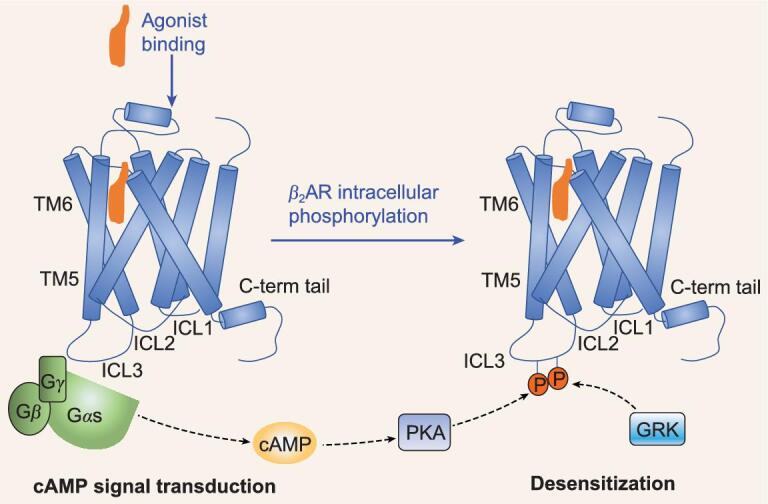
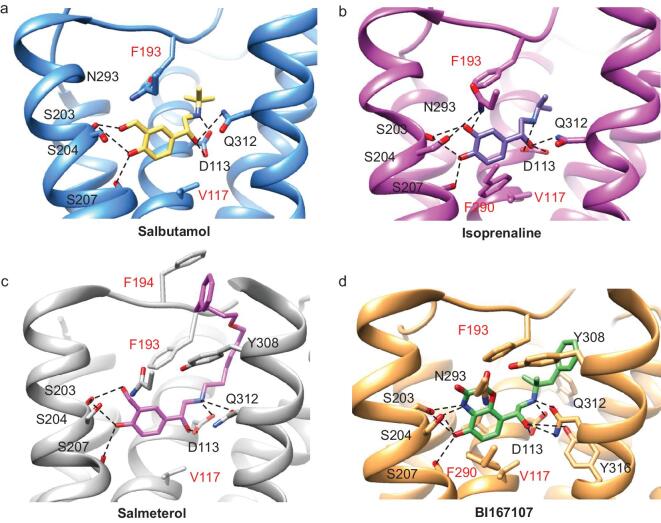
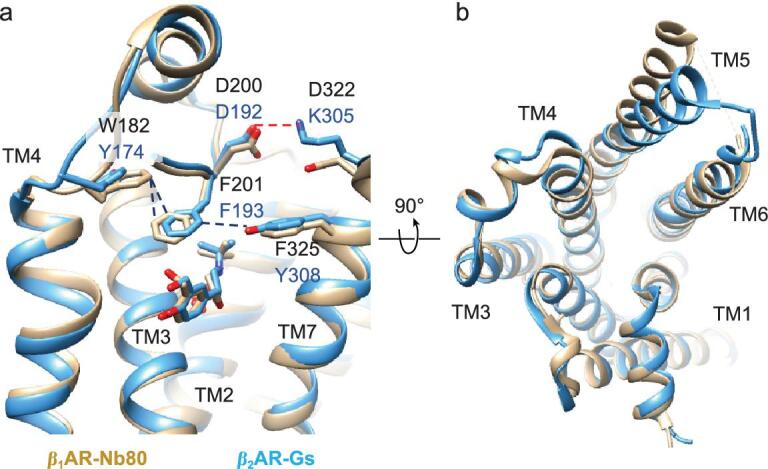
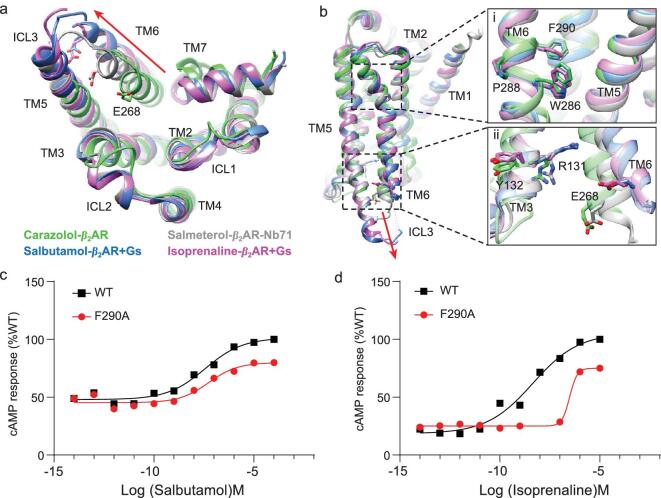
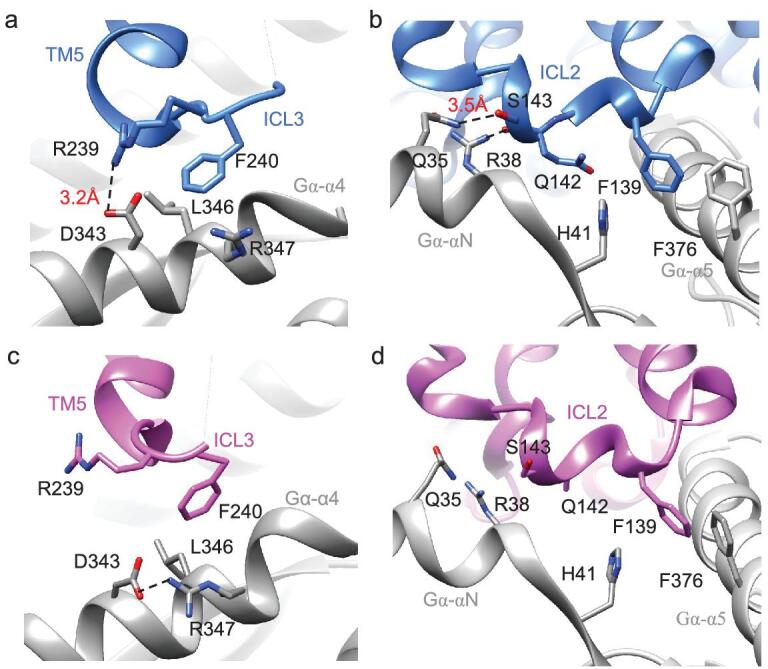
G protein-coupled receptors (GPCRs) are responsible for most cytoplasmic signaling in response to extracellular ligands with different efficacy profiles. Various spectroscopic techniques have identified that agonists exhibiting varying efficacies can selectively stabilize a specific conformation of the receptor. However, the structural basis for activation of the GPCR-G protein complex by ligands with different efficacies is incompletely understood. To better understand the structural basis underlying the mechanisms by which ligands with varying efficacies differentially regulate the conformations of receptors and G proteins, we determined the structures of βAR-Gα[Formula: see text]γ bound with partial agonist salbutamol or bound with full agonist isoprenaline using single-particle cryo-electron microscopy at resolutions of 3.26 Å and 3.80 Å, respectively. Structural comparisons between the βAR-Gs-salbutamol and βAR-Gs-isoprenaline complexes demonstrated that the decreased binding affinity and efficacy of salbutamol compared with those of isoprenaline might be attributed to weakened hydrogen bonding interactions, attenuated hydrophobic interactions in the orthosteric binding pocket and different conformational changes in the rotamer toggle switch in TM6. Moreover, the observed stronger interactions between the intracellular loop 2 or 3 (ICL2 or ICL3) of βAR and Gα with binding of salbutamol versus isoprenaline might decrease phosphorylation in the salbutamol-activated βAR-Gs complex. From the observed structural differences between these complexes of βAR, a mechanism of βAR activation by partial and full agonists is proposed to provide structural insights into βAR desensitization.
G蛋白偶联受体(GPCRs)负责大多数细胞内信号传导,以响应具有不同效能特征的细胞外配体。各种光谱技术已确定,具有不同效能的激动剂可选择性地稳定受体的特定构象。然而,不同效能的配体激活GPCR-G蛋白复合物的结构基础仍未完全了解。为了更好地理解不同效能的配体差异调节受体和G蛋白构象的机制背后的结构基础,我们分别使用单颗粒冷冻电子显微镜在3.26 Å和3.80 Å的分辨率下,确定了与部分激动剂沙丁胺醇结合或与完全激动剂异丙肾上腺素结合的βAR-Gα[公式:见正文]γ的结构。βAR-Gs-沙丁胺醇和βAR-Gs-异丙肾上腺素复合物之间的结构比较表明,与异丙肾上腺素相比,沙丁胺醇结合亲和力和效能的降低可能归因于氢键相互作用减弱、正构结合口袋中疏水相互作用减弱以及TM6中旋转异构体切换开关的不同构象变化。此外,观察到与异丙肾上腺素结合相比,沙丁胺醇结合时βAR的细胞内环2或3(ICL2或ICL3)与Gα之间更强的相互作用可能会降低沙丁胺醇激活的βAR-Gs复合物中的磷酸化。从这些βAR复合物之间观察到的结构差异,提出了部分激动剂和完全激动剂激活βAR的机制,以提供对βAR脱敏的结构见解。