Haines Jeremiah, Garster Noelle, Mohananey Divyanshu, Safarova Maya S
Division of Cardiovascular Medicine, Department of Medicine, Medical College of Wisconsin, 8701 W Watertown Plank Rd, Milwaukee, WI 53226, USA.
Eur Heart J Case Rep. 2024 Jul 18;8(7):ytae321. doi: 10.1093/ehjcr/ytae321. eCollection 2024 Jul.
Arrhythmogenic cardiomyopathy (ACM) is a genetically determined myocardial atrophy which progressively extends from the epicardium towards the endocardium, resulting in wall thinning. It is one of the leading causes of sudden death in young people. Postmortem studies demonstrate that up to 70-80% of the cases have biventricular involvement. Variable penetrance and expressivity results in a wide phenotypic spectrum, challenging diagnostic accuracy of advanced multimodality imaging tools. Prompt recognition, non-invasive imaging, risk stratification for sudden cardiac death (SCD), and preventive measures are paramount to improve prognosis.
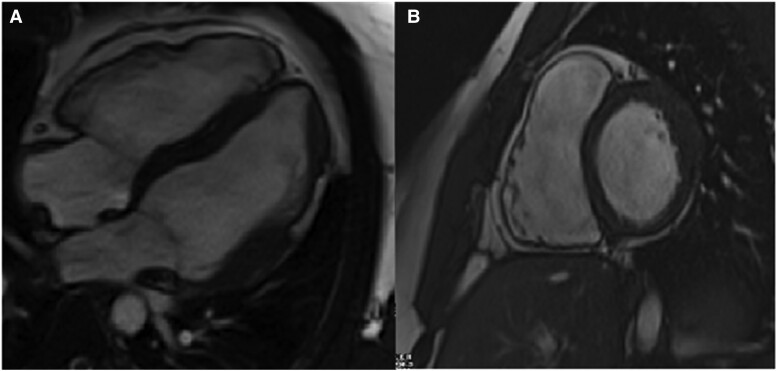
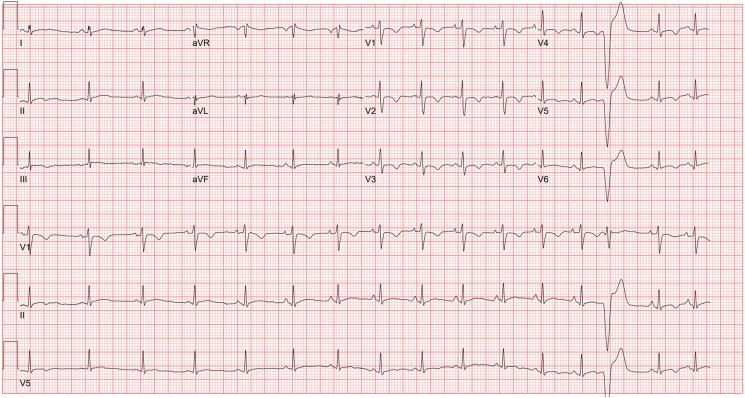
Here, we present a 22-year-old Black male who was referred to our electrophysiology clinic with palpitations, remote syncope, and a family history of SCD. Over 3 years, he developed gradually worsening symptomatic palpitations. While physical exam and transthoracic echocardiography were unremarkable, his cardiac magnetic resonance imaging was consistent with biventricular ACM. Genetic testing confirmed ACM, revealing double heterozygosity in and . Given the elevated estimated risk of life-threatening dysrhythmias, a subcutaneous cardiac defibrillator was successfully implanted.
Frequently, patients with ACM have more than one mutation in the same gene (compound heterozygosity) or in a second gene (double heterozygosity). Genetic counselling is strongly recommended for family members of the proband. The diagnosis of ACM may be mimicked by other diseases (cardiac sarcoidosis, dilated cardiomyopathy, amyloidosis), thus genetic testing can be useful to determine the presence of the disease. The present report provides an overview of the clinical course, diagnostic criteria, risk stratification, and prognostication for patients with ACM.
致心律失常性心肌病(ACM)是一种由基因决定的心肌萎缩疾病,其病变从心外膜逐渐向心内膜扩展,导致心室壁变薄。它是年轻人猝死的主要原因之一。尸检研究表明,高达70%-80%的病例存在双心室受累。可变的外显率和表现度导致了广泛的表型谱,对先进的多模态成像工具的诊断准确性提出了挑战。及时识别、无创成像、心脏性猝死(SCD)风险分层以及预防措施对于改善预后至关重要。
在此,我们报告一名22岁的黑人男性,因心悸、既往晕厥以及SCD家族史被转诊至我们的电生理门诊。在3年多的时间里,他的症状性心悸逐渐加重。体格检查和经胸超声心动图检查均无异常,但他的心脏磁共振成像结果符合双心室ACM。基因检测确诊为ACM,显示在[具体基因1]和[具体基因2]存在双重杂合性。鉴于危及生命的心律失常风险估计值升高,成功植入了皮下心脏除颤器。
通常,ACM患者在同一基因中存在不止一个突变(复合杂合性)或在第二个基因中存在突变(双重杂合性)。强烈建议为先证者的家庭成员提供遗传咨询。ACM的诊断可能会被其他疾病(心脏结节病、扩张型心肌病、淀粉样变性)所模仿,因此基因检测有助于确定疾病的存在。本报告概述了ACM患者的临床病程、诊断标准、风险分层和预后情况。