Department of Biomedical Engineering, University of Iowa, Iowa City, Iowa, United States.
Department of Internal Medicine, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, United States.
Am J Physiol Lung Cell Mol Physiol. 2024 Oct 1;327(4):L415-L422. doi: 10.1152/ajplung.00010.2024. Epub 2024 Aug 6.
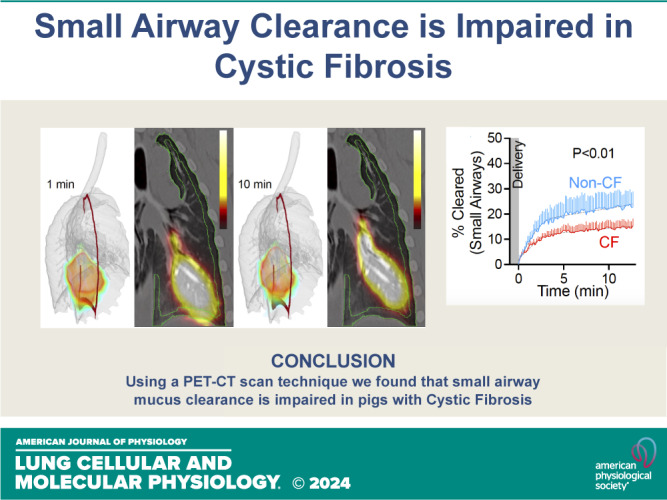
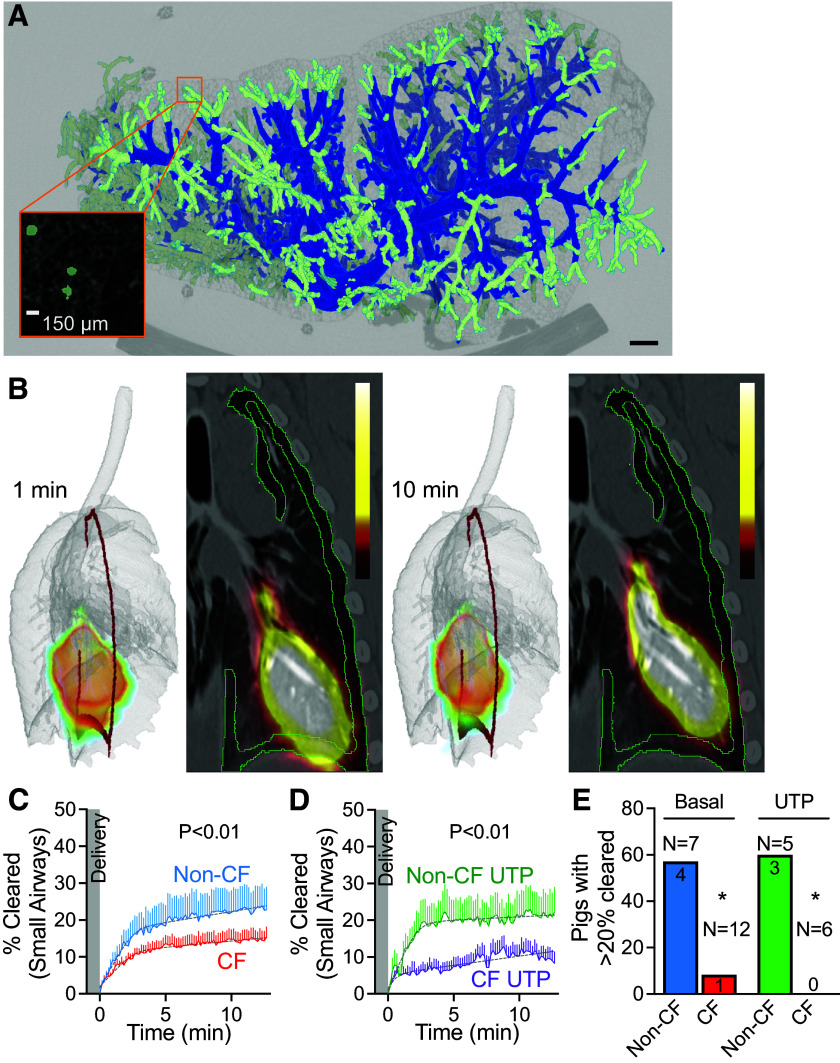
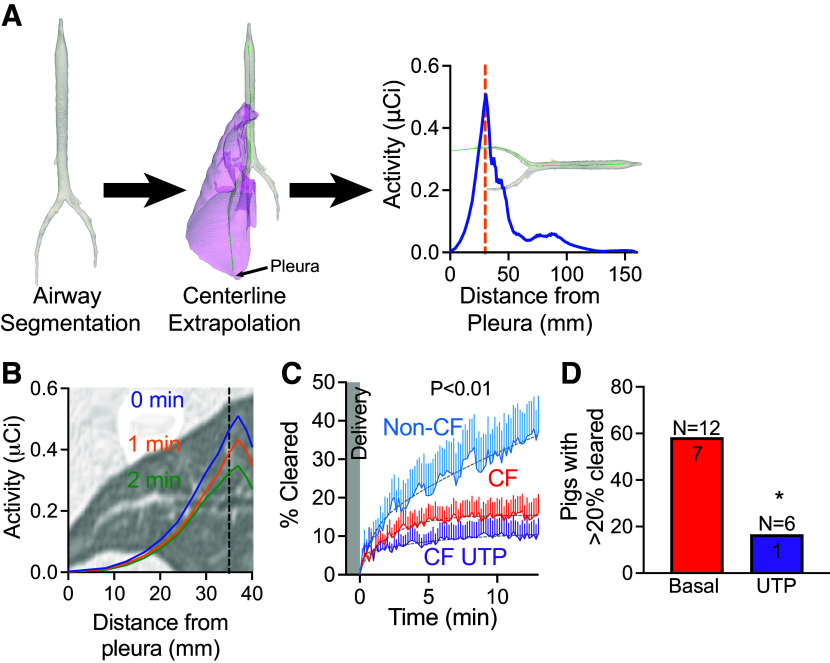
Cystic fibrosis (CF) is a genetic disorder characterized by recurrent airway infections, inflammation, impaired mucociliary clearance, and progressive decline in lung function. The disease may start in the small airways; however, this is difficult to prove due to the limited accessibility of the small airways with the current single-photon mucociliary clearance assay. Here, we developed a dynamic positron emission tomography assay with high spatial and temporal resolution. We tested that mucociliary clearance is abnormal in the small airways of newborn cystic fibrosis pigs. Clearance of [Ga]-tagged macroaggregated albumin from small airways started immediately after delivery and continued for the duration of the study. Initial clearance was fast but slowed down a few minutes after delivery. Cystic fibrosis pigs' small airways cleared significantly less than non-CF pigs' small airways (non-CF 25.1 ± 3.1% vs. CF 14.6 ± 0.1%). Stimulation of the cystic fibrosis airways with the purinergic secretagogue uridine-5'-triphosphate (UTP) further impaired clearance (non-CF with UTP 20.9 ± 0.3% vs. CF with UTP 13.0 ± 1.8%). None of the cystic fibrosis pigs treated with UTP ( = 6) cleared more than 20% of the delivered dose. These data indicate that mucociliary clearance in the small airways is fast and can easily be missed if the assay is not sensitive enough. The data also indicate that mucociliary clearance is impaired in the small airways of cystic fibrosis pigs. This defect is exacerbated by stimulation of mucus secretions with purinergic agonists. We developed a novel positron emission tomography scan assay with unprecedented temporal and spatial resolution to measure mucociliary clearance in the small airways. We proved a long-standing but unproven assertion that mucociliary clearance is inherently abnormal in the small airways of newborn cystic fibrosis piglets that are otherwise free of infection or inflammation. This technique can be easily extended to other airway diseases such as asthma, idiopathic pulmonary fibrosis, or chronic obstructive pulmonary disease.
囊性纤维化 (CF) 是一种遗传性疾病,其特征是反复发生呼吸道感染、炎症、黏液纤毛清除功能受损以及肺功能进行性下降。该疾病可能首先发生在小气道中;然而,由于当前的单光子黏液纤毛清除测定法对小气道的有限可及性,这很难得到证明。在这里,我们开发了一种具有高时空分辨率的动态正电子发射断层扫描测定法。我们测试了新生 CF 猪的小气道黏液纤毛清除功能异常。[Ga]标记的大聚合白蛋白从小气道中的清除在出生后立即开始,并持续到研究结束。最初的清除速度很快,但在出生后几分钟后会减慢。CF 猪的小气道清除率明显低于非 CF 猪的小气道清除率(非 CF 为 25.1 ± 3.1%,CF 为 14.6 ± 0.1%)。用嘌呤能分泌激动剂尿苷-5'-三磷酸 (UTP) 刺激 CF 气道进一步损害了清除率(非 CF 加 UTP 为 20.9 ± 0.3%,CF 加 UTP 为 13.0 ± 1.8%)。没有一只接受 UTP 治疗的 CF 猪(n = 6)的清除率超过输送剂量的 20%。这些数据表明,小气道中的黏液纤毛清除速度很快,如果测定法不够灵敏,很容易错过。数据还表明,CF 猪的小气道黏液纤毛清除功能受损。用嘌呤能激动剂刺激黏液分泌会加剧这种缺陷。我们开发了一种新的正电子发射断层扫描扫描测定法,具有前所未有的时间和空间分辨率,用于测量小气道中的黏液纤毛清除率。我们证明了一个长期以来但未经证实的断言,即在没有感染或炎症的情况下,新生 CF 仔猪的小气道中固有地存在黏液纤毛清除异常。该技术可以很容易地扩展到其他气道疾病,如哮喘、特发性肺纤维化或慢性阻塞性肺疾病。