Xu Jie, Li Ruihan, Ma Yijian, Zhu Jie, Shen Chengshuo, Jiang Heng
Shanghai key Laboratory for Molecular Engineering of Chiral Drugs, Shanghai Frontiers Science Center of Drug Target Identification and Delivery, School of Pharmaceutical Sciences, Shanghai Jiao Tong University, Shanghai, China.
School of Chemistry and Chemical Engineering, Zhejiang Sci-Tech University, Hangzhou, China.
Nat Commun. 2024 Aug 9;15(1):6791. doi: 10.1038/s41467-024-51239-3.
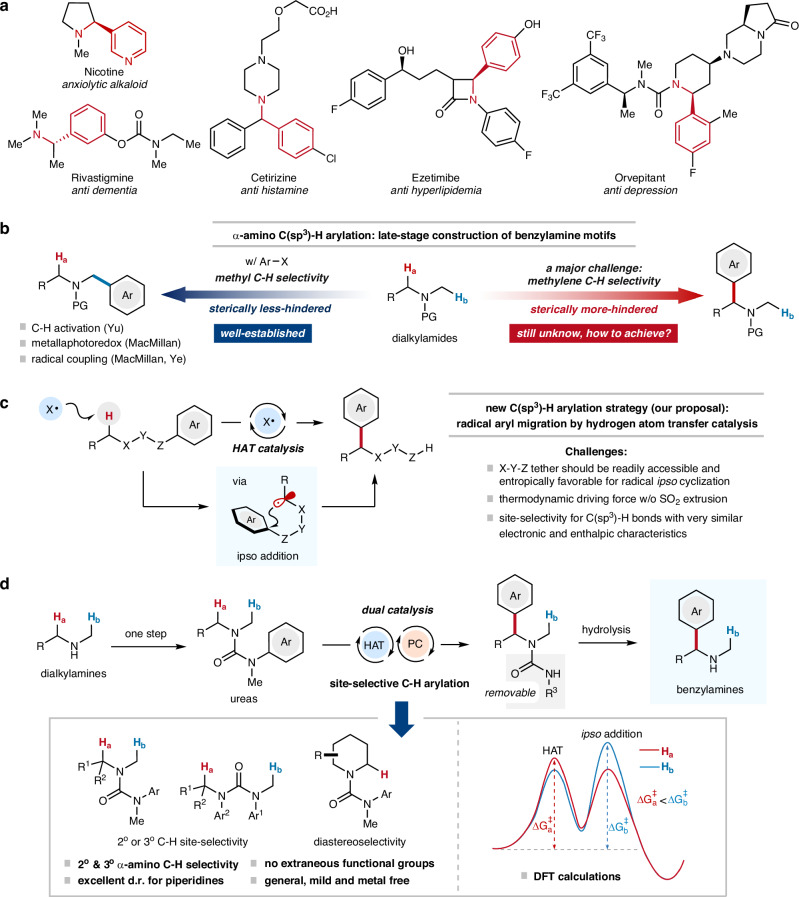
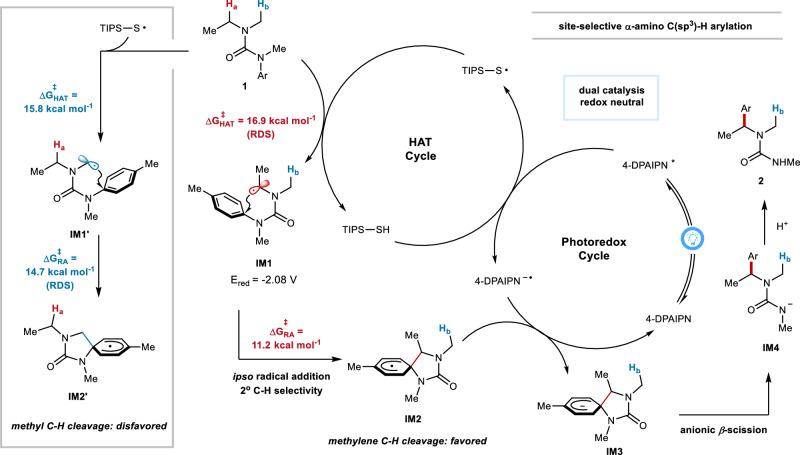
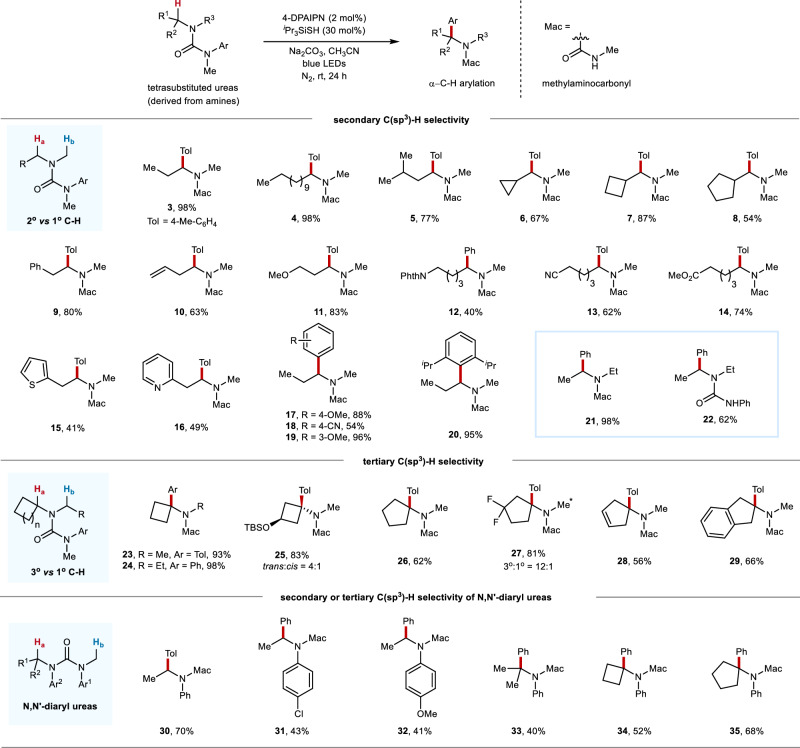
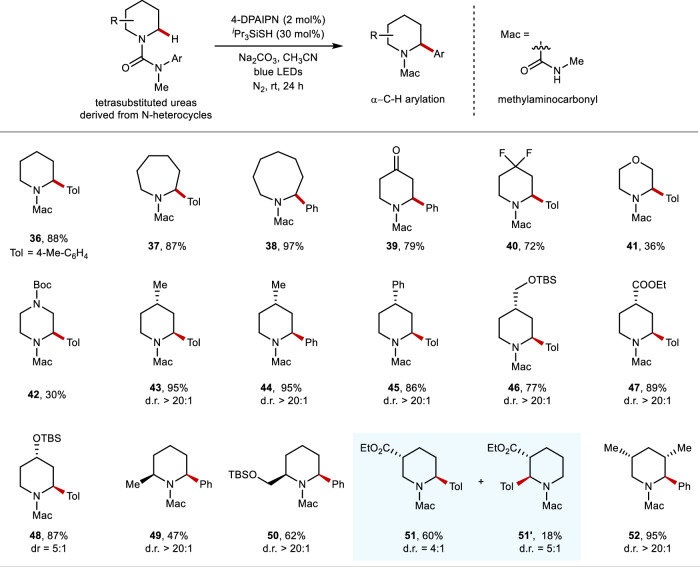
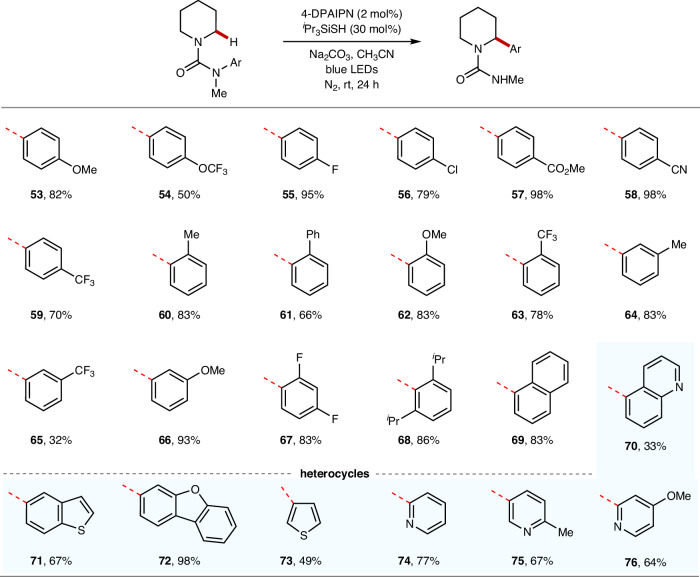
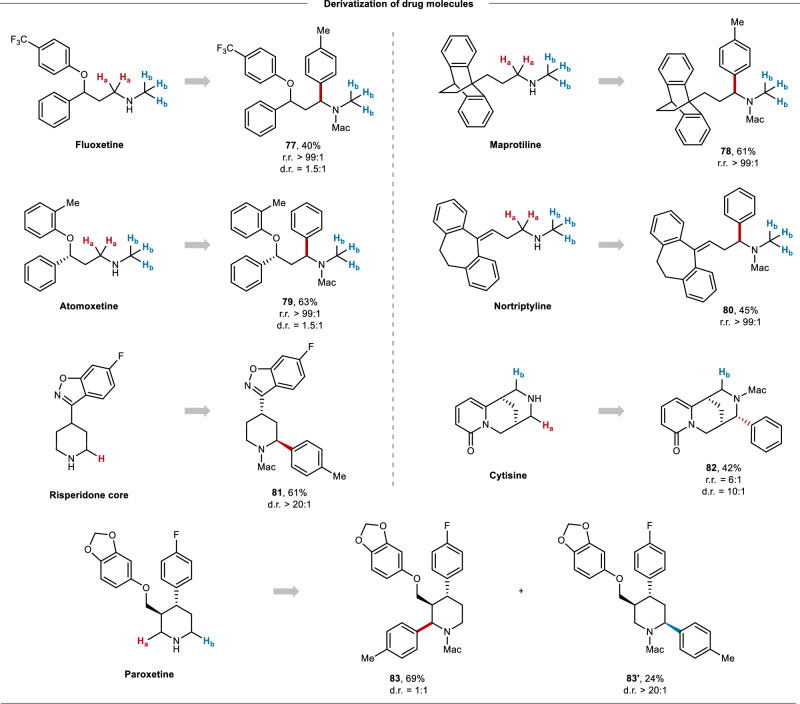
Site-selective C(sp)-H arylation is an appealing strategy to synthesize complex arene structures but remains a challenge facing synthetic chemists. Here we report the use of photoredox-mediated hydrogen atom transfer (HAT) catalysis to accomplish the site-selective α-C(sp)-H arylation of dialkylamine-derived ureas through 1,4-radical aryl migration, by which a wide array of benzylamine motifs can be incorporated to the medicinally relevant systems in the late-stage installation steps. In contrast to previous efforts, this C-H arylation protocol exhibits specific site-selectivity, proforming predominantly on sterically more-hindered secondary and tertiary α-amino carbon centers, while the C-H functionalization of sterically less-hindered N-methyl group can be effectively circumvented in most cases. Moreover, a diverse range of multi-substituted piperidine derivatives can be obtained with excellent diastereoselectivity. Mechanistic and computational studies demonstrate that the rate-determining step for methylene C-H arylation is the initial H atom abstraction, whereas the radical ipso cyclization step bears the highest energy barrier for N-methyl functionalization. The relatively lower activation free energies for secondary and tertiary α-amino C-H arylation compared with the functionalization of methylic C-H bond lead to the exceptional site-selectivity.
位点选择性C(sp)-H芳基化是合成复杂芳烃结构的一种有吸引力的策略,但仍然是合成化学家面临的一个挑战。在此,我们报道了利用光氧化还原介导的氢原子转移(HAT)催化,通过1,4-自由基芳基迁移实现二烷基胺衍生脲的位点选择性α-C(sp)-H芳基化,通过该方法可以在后期安装步骤中将多种苄胺基序引入到与药物相关的体系中。与之前的研究不同,这种C-H芳基化方案表现出特定的位点选择性,主要在空间位阻较大的仲和叔α-氨基碳中心进行,而在大多数情况下,可以有效避免空间位阻较小的N-甲基的C-H官能化。此外,可以以优异的非对映选择性获得多种多取代哌啶衍生物。机理和计算研究表明,亚甲基C-H芳基化的决速步骤是最初的氢原子提取,而自由基本位环化步骤是N-甲基官能化的最高能量屏障。与甲基C-H键的官能化相比,仲和叔α-氨基C-H芳基化相对较低的活化自由能导致了优异的位点选择性。