Department of Chemistry, Columbia University, New York, New York 10027, United States.
Institute of Organic Chemistry, RWTH Aachen University, 52047 Aachen, Germany.
J Am Chem Soc. 2021 Nov 17;143(45):18952-18959. doi: 10.1021/jacs.1c07144. Epub 2021 Nov 5.
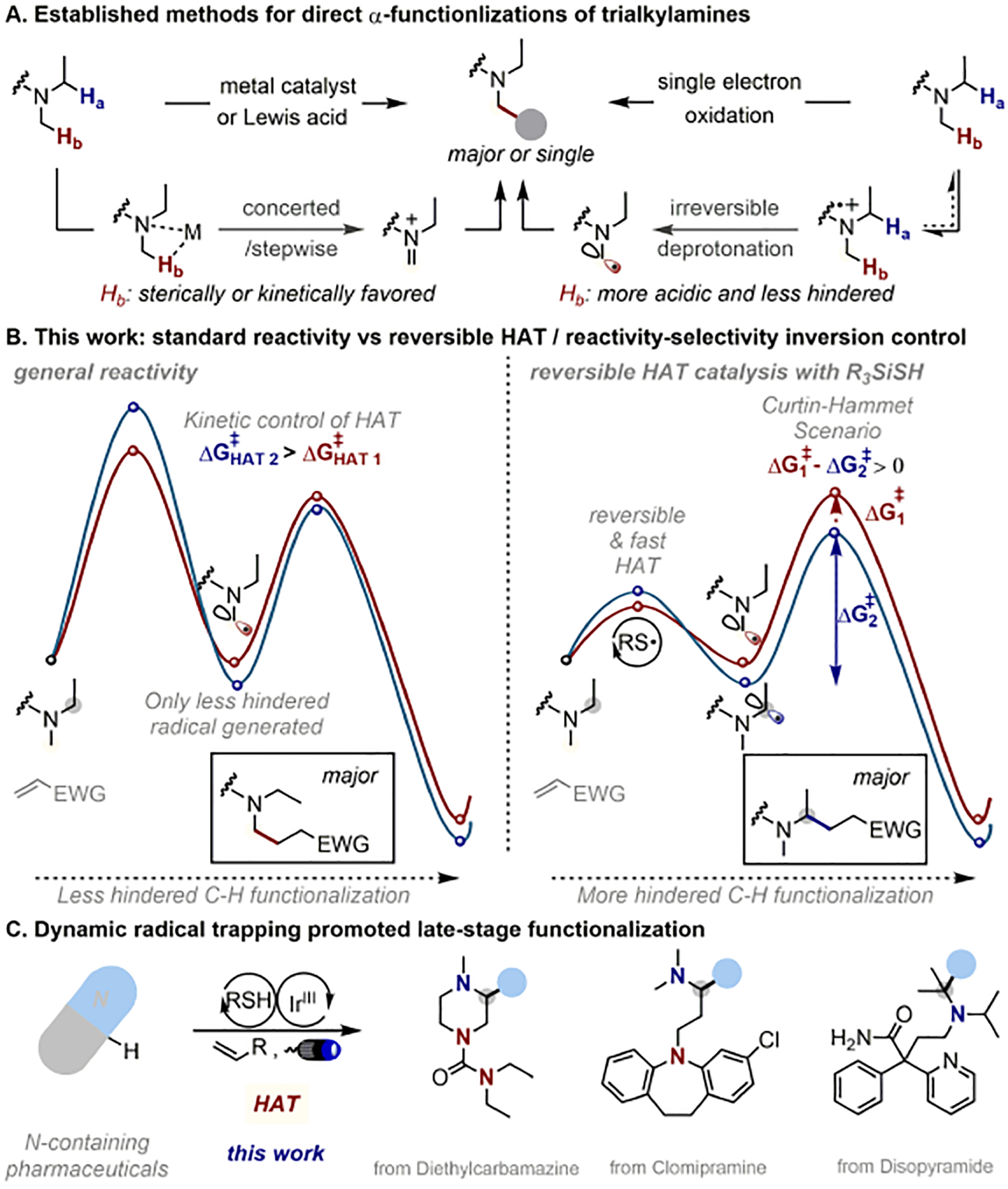
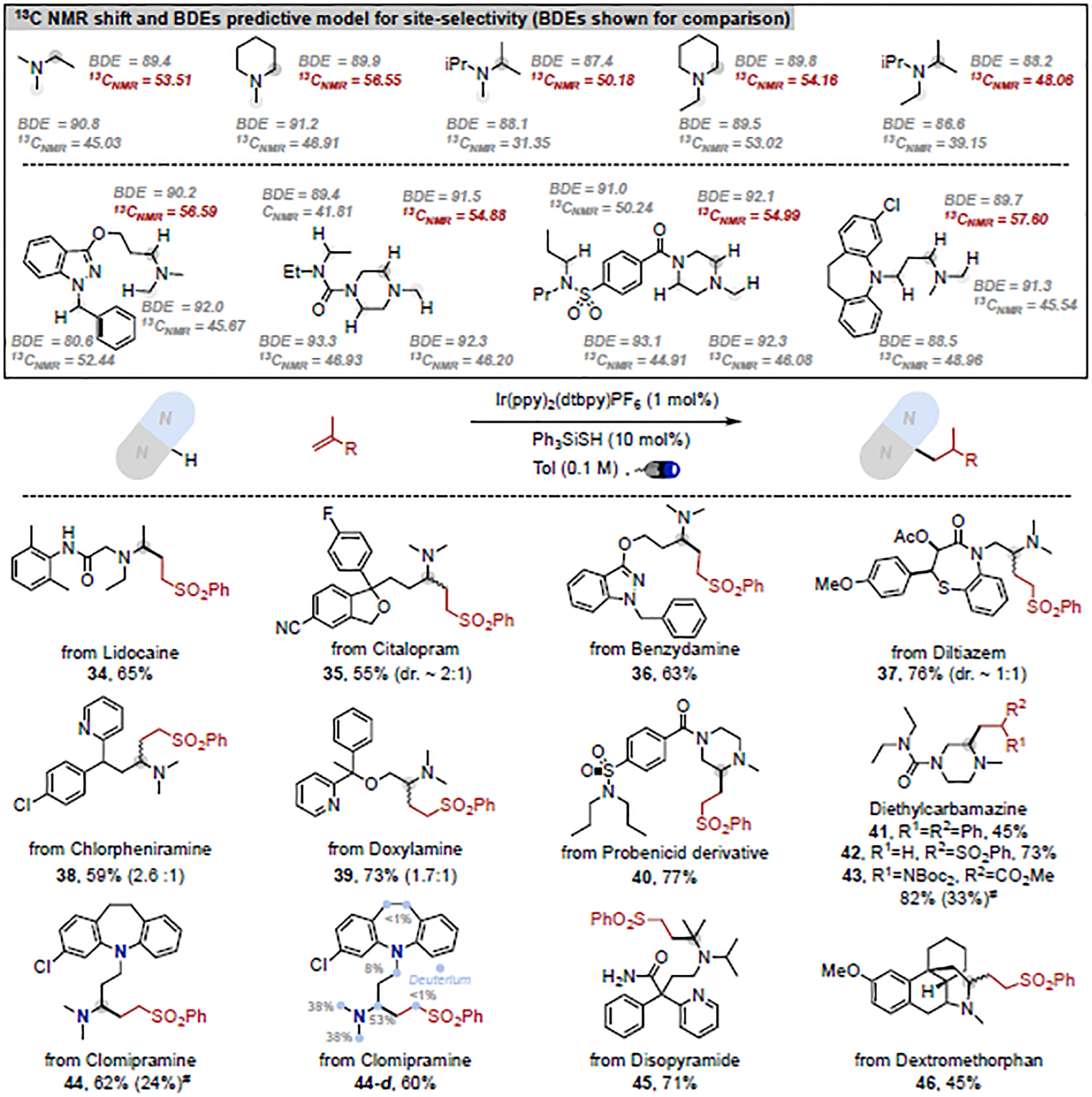
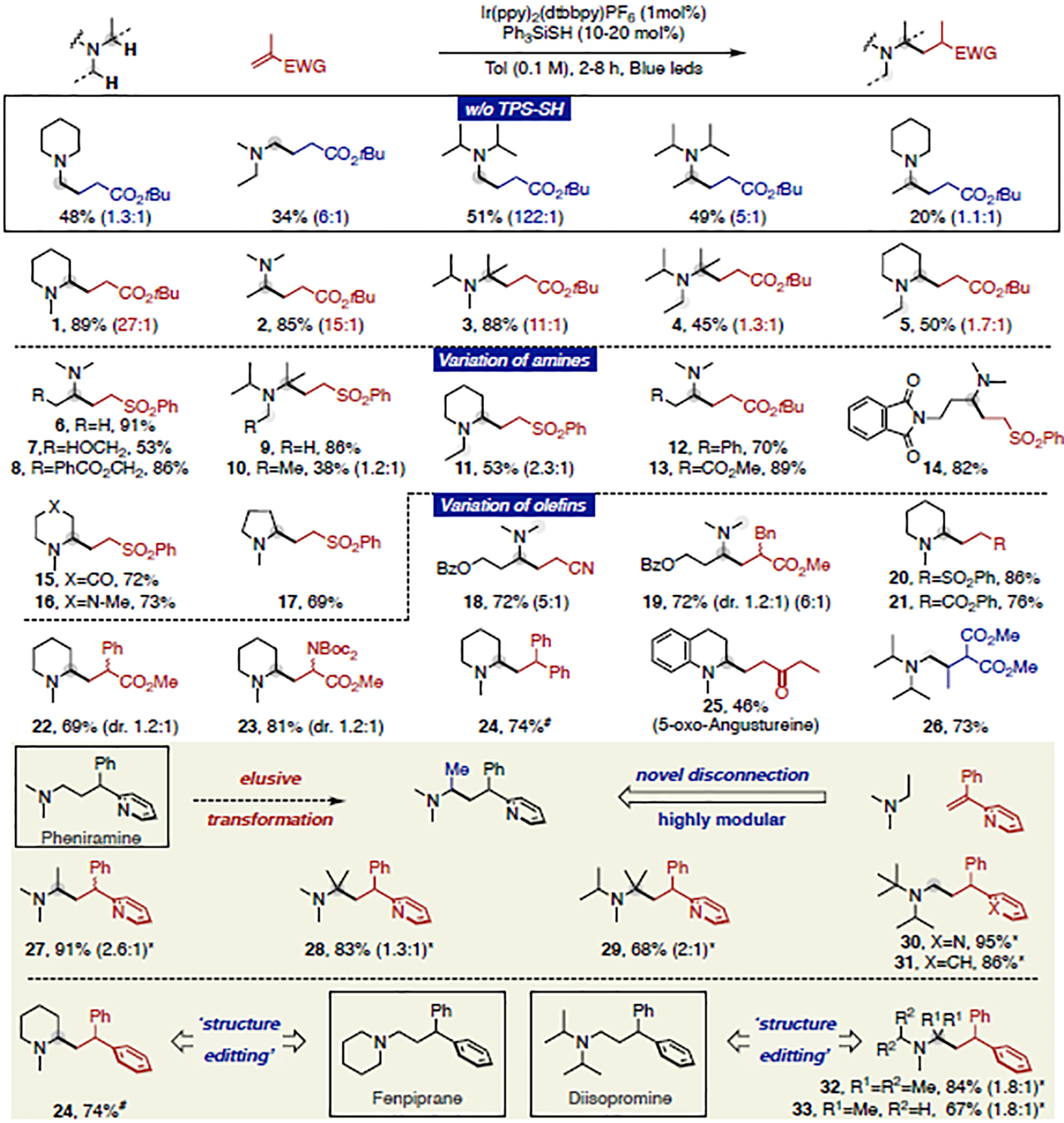
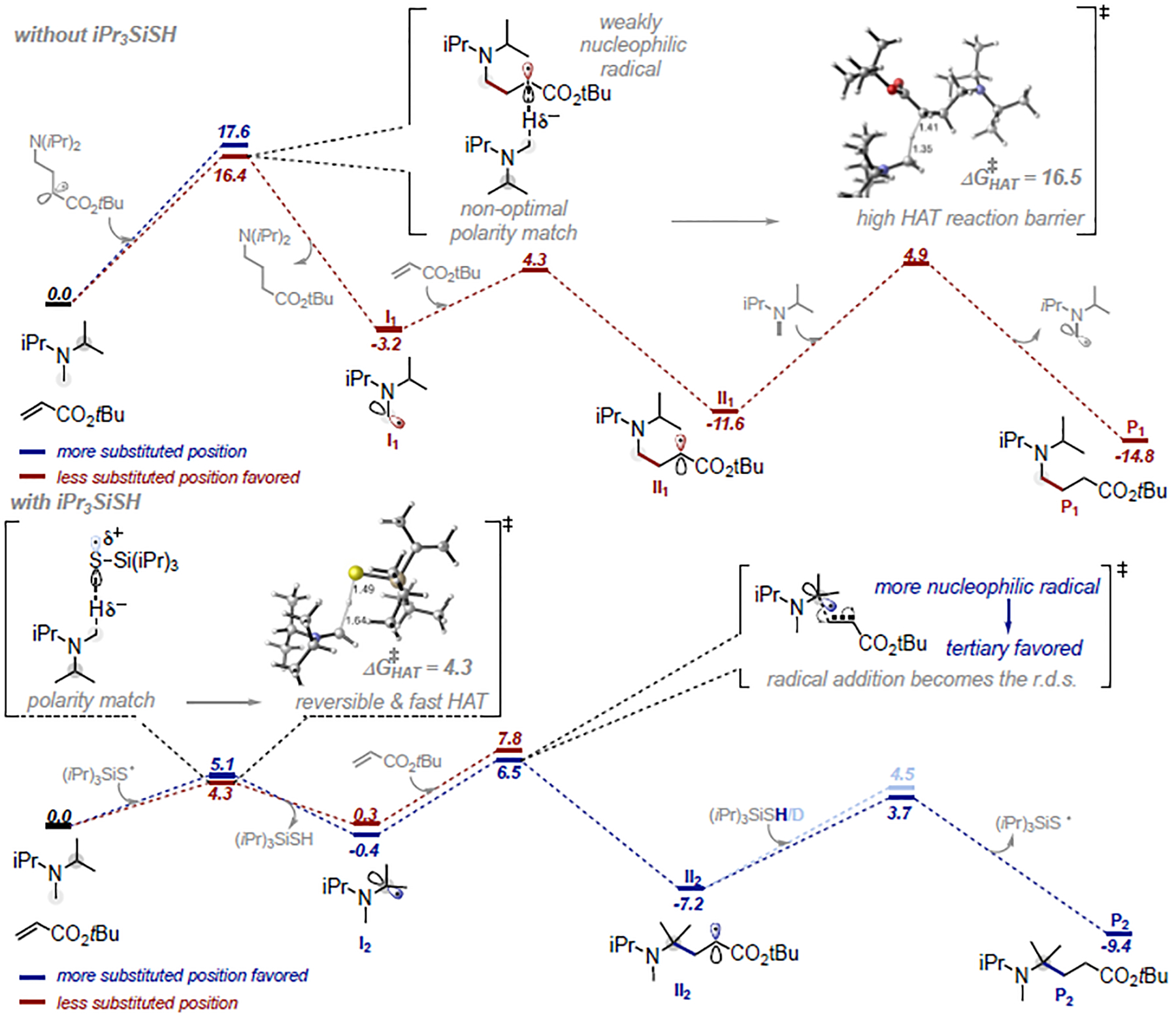
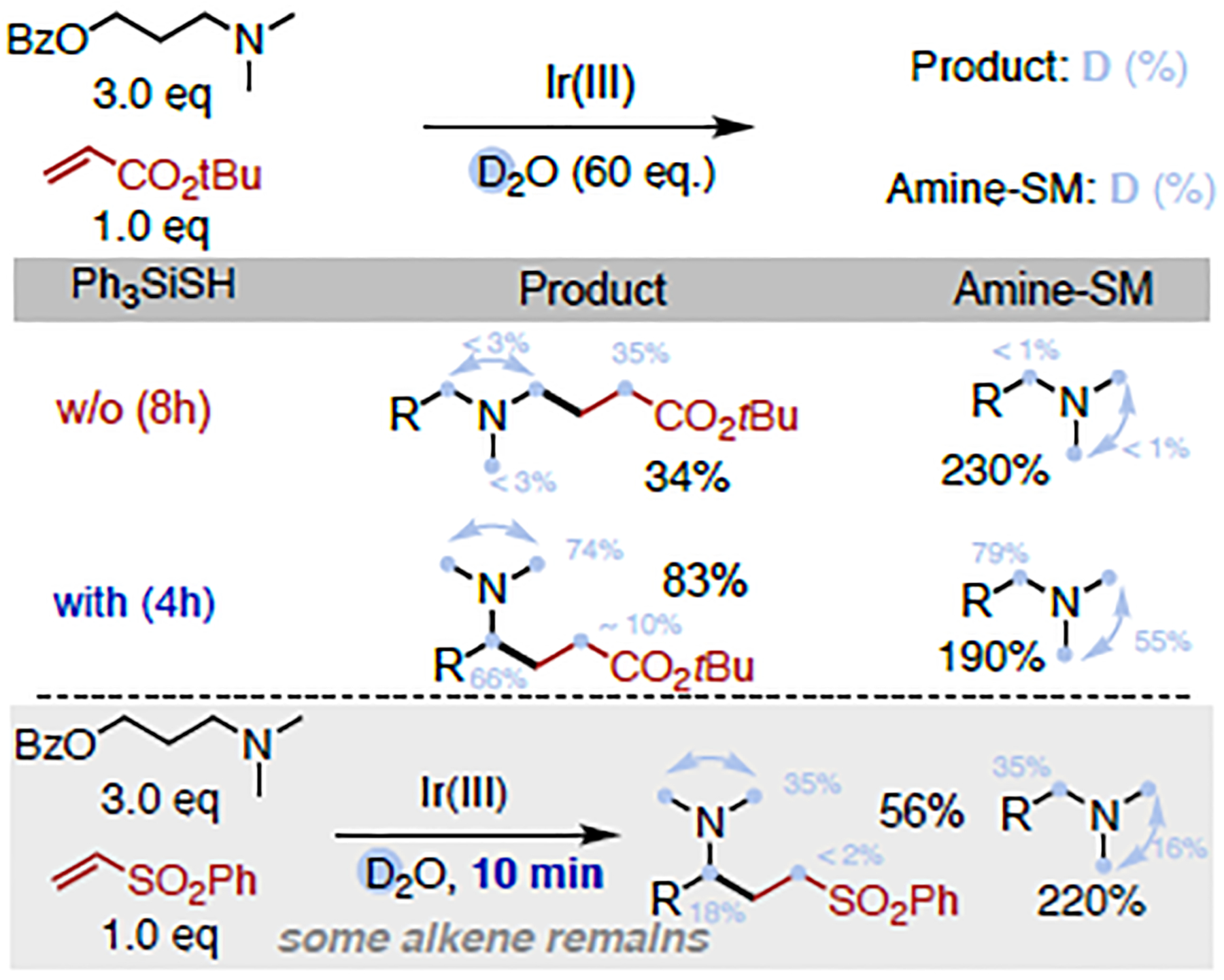
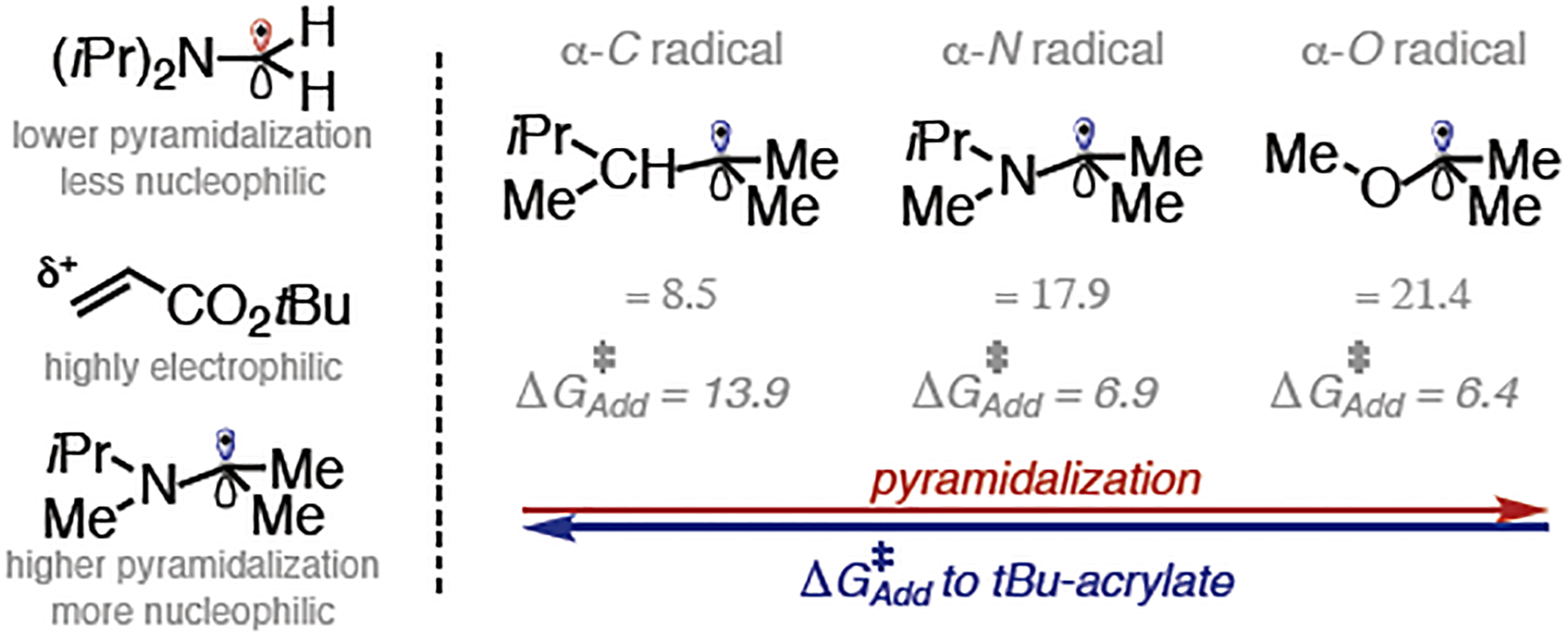
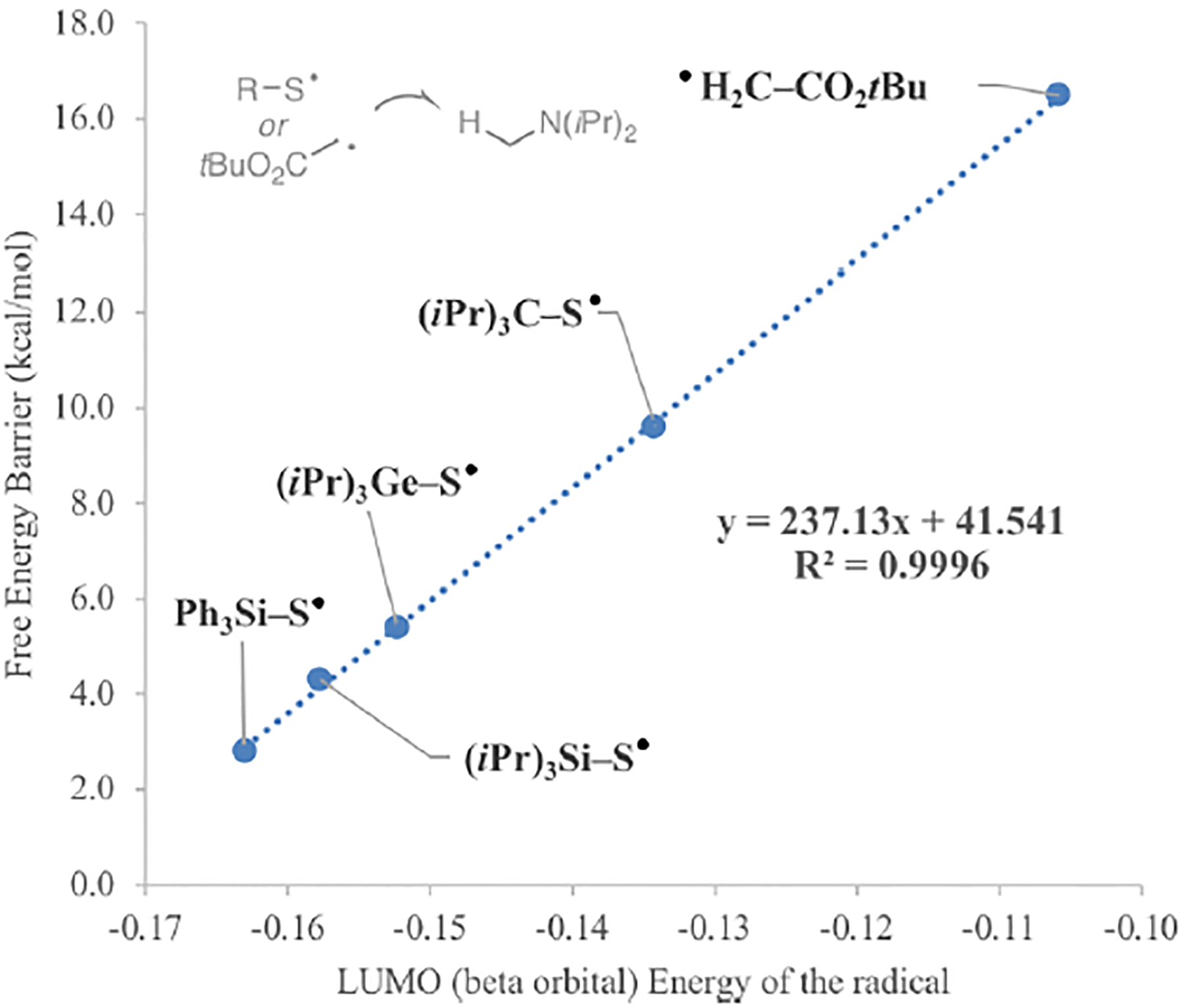
Trialkylamines are widely found in naturally occurring alkaloids, synthetic agrochemicals, biological probes, and especially pharmaceuticals agents and preclinical candidates. Despite the recent breakthrough of catalytic alkylation of dialkylamines, the selective α-C(sp)-H bond functionalization of widely available trialkylamine scaffolds holds promise to streamline complex trialkylamine synthesis, accelerate drug discovery, and execute late-stage pharmaceutical modification with complementary reactivity. However, the canonical methods always result in functionalization at the less-crowded site. Herein, we describe a solution to switch the reaction site through fundamentally overcoming the steric control that dominates such processes. By rapidly establishing an equilibrium between α-amino C(sp)-H bonds and a highly electrophilic thiol radical via reversible hydrogen atom transfer, we leverage a slower radical-trapping step with electron-deficient olefins to selectively forge a C(sp)-C(sp) bond with the more-crowded α-amino radical, with the overall selectivity guided by the Curtin-Hammett principle. This subtle reaction profile has unlocked a new strategic concept in direct C-H functionalization arena for forging C-C bonds from a diverse set of trialkylamines with high levels of site selectivity and preparative utility. Simple correlation of site selectivity and C NMR shift serves as a qualitative predictive guide. The broad consequences of this dynamic system, together with the ability to forge N-substituted quaternary carbon centers and implement late-stage functionalization techniques, hold potential to streamline complex trialkylamine synthesis and accelerate small-molecule drug discovery.
三烷基胺广泛存在于天然存在的生物碱、合成农用化学品、生物探针中,特别是在药物制剂和临床前候选药物中。尽管最近在二烷基胺的催化烷基化方面取得了突破,但广泛可用的三烷基胺支架的选择性α-C(sp)-H 键功能化有望简化复杂的三烷基胺合成、加速药物发现,并通过互补的反应性执行晚期药物修饰。然而,典型的方法总是导致在较不拥挤的位置进行官能化。在这里,我们通过从根本上克服主导这些过程的空间位阻控制来描述一种改变反应位点的解决方案。通过快速通过可逆氢原子转移在α-氨基 C(sp)-H 键和高亲电性硫自由基之间建立平衡,我们利用较慢的自由基捕获步骤与缺电子烯烃反应,选择性地在更拥挤的α-氨基自由基上形成 C(sp)-C(sp)键,总体选择性受 Curtin-Hammett 原理指导。这种微妙的反应轮廓为直接 C-H 官能化领域解锁了一个新的战略概念,用于从各种三烷基胺中形成 C-C 键,具有高的位点选择性和可制备性。位点选择性和 C NMR 位移的简单相关性可用作定性预测指南。这种动态系统的广泛影响,以及形成 N-取代的季碳原子中心和实施晚期功能化技术的能力,有可能简化复杂的三烷基胺合成并加速小分子药物发现。