Liu Chao, Wu Pei, Wu Xue, Zhao Xia, Chen Fang, Cheng Xiaofang, Zhu Hongmei, Wang Ou, Xu Mengyang
BGI, Tianjin, China.
BGI Research, Shenzhen, China.
Front Genet. 2024 Jul 26;15:1421565. doi: 10.3389/fgene.2024.1421565. eCollection 2024.
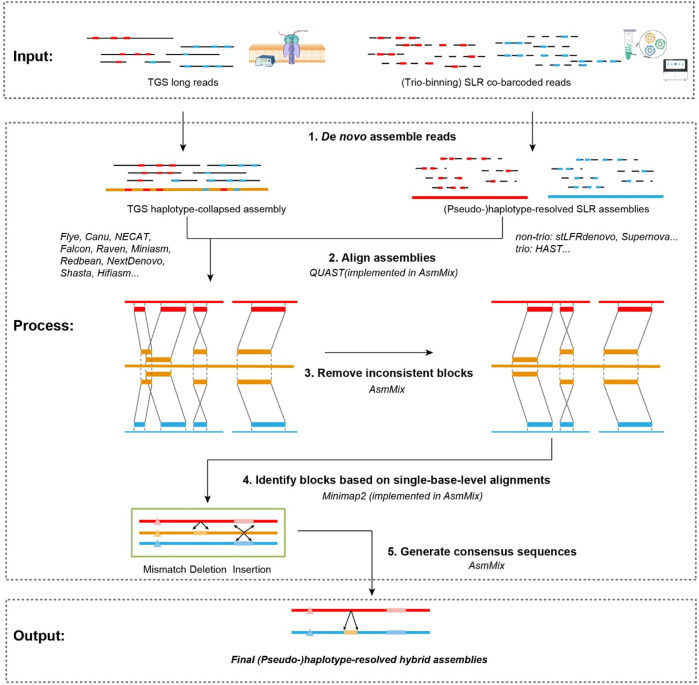
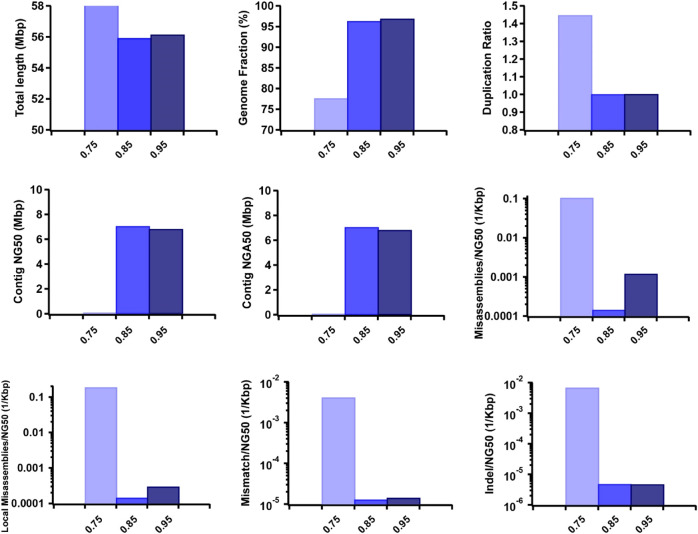
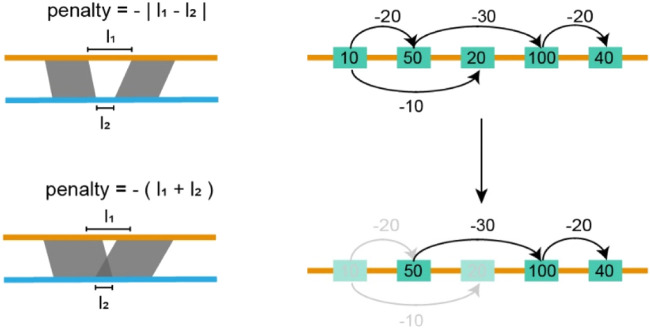
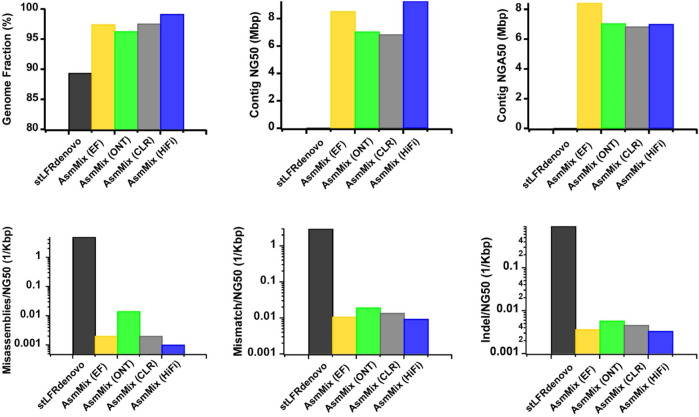
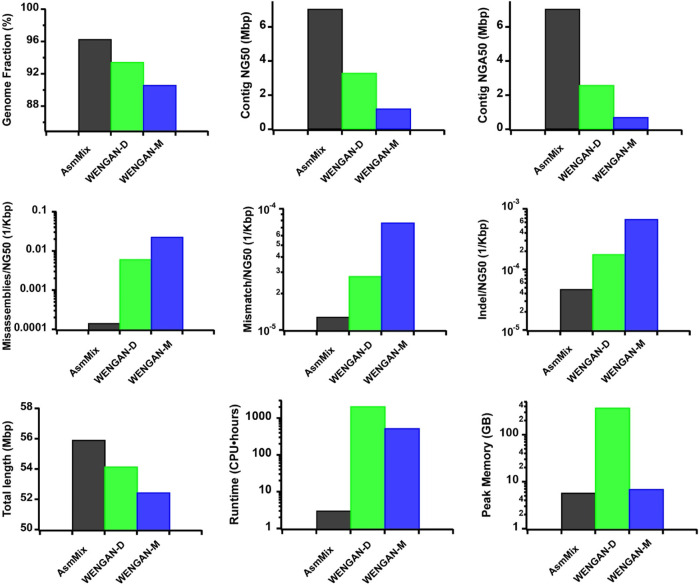
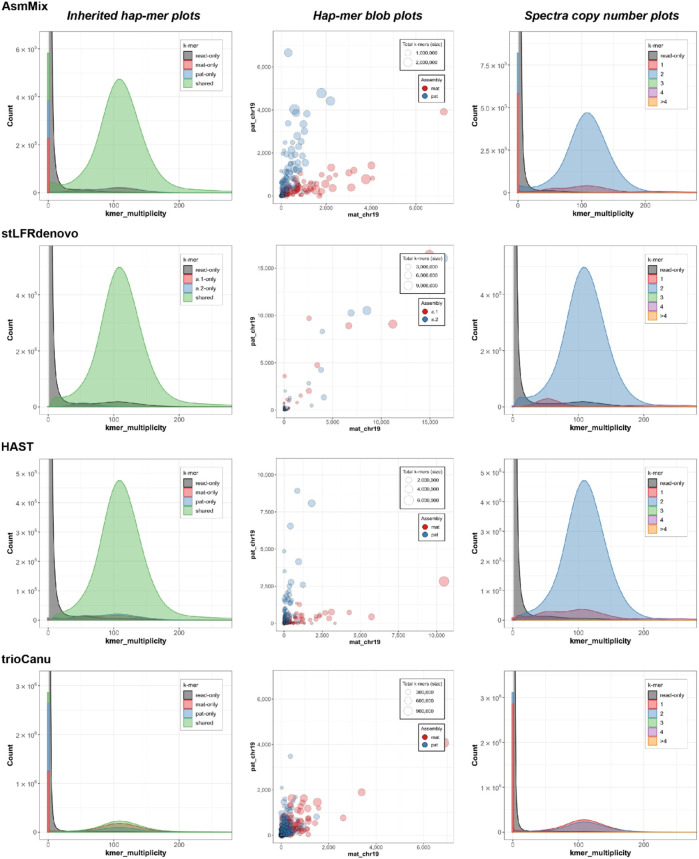
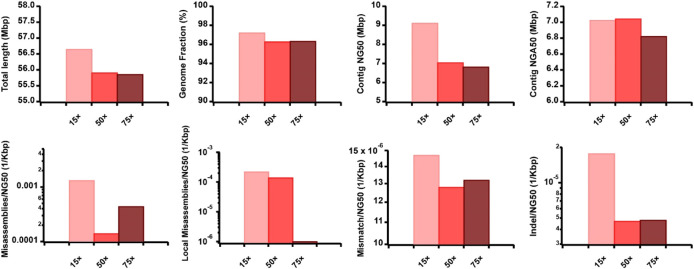
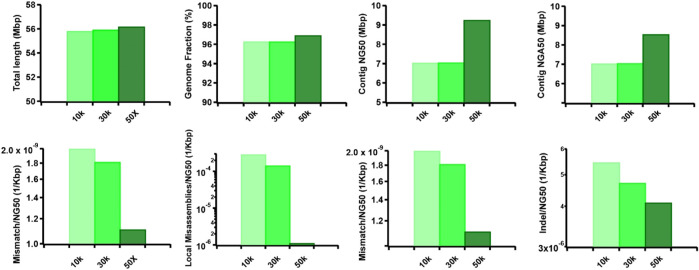
Accurate haplotyping facilitates distinguishing allele-specific expression, identifying cis-regulatory elements, and characterizing genomic variations, which enables more precise investigations into the relationship between genotype and phenotype. Recent advances in third-generation single-molecule long read and synthetic co-barcoded read sequencing techniques have harnessed long-range information to simplify the assembly graph and improve assembly genomic sequence. However, it remains methodologically challenging to reconstruct the complete haplotypes due to high sequencing error rates of long reads and limited capturing efficiency of co-barcoded reads. We here present a pipeline, AsmMix, for generating both contiguous and accurate diploid genomes. It first assembles co-barcoded reads to generate accurate haplotype-resolved assemblies that may contain many gaps, while the long-read assembly is contiguous but susceptible to errors. Then two assembly sets are integrated into haplotype-resolved assemblies with reduced misassembles. Through extensive evaluation on multiple synthetic datasets, AsmMix consistently demonstrates high precision and recall rates for haplotyping across diverse sequencing platforms, coverage depths, read lengths, and read accuracies, significantly outperforming other existing tools in the field. Furthermore, we validate the effectiveness of our pipeline using a human whole genome dataset (HG002), and produce highly contiguous, accurate, and haplotype-resolved assemblies. These assemblies are evaluated using the GIAB benchmarks, confirming the accuracy of variant calling. Our results demonstrate that AsmMix offers a straightforward yet highly efficient approach that effectively leverages both long reads and co-barcoded reads for haplotype-resolved assembly.
准确的单倍型分型有助于区分等位基因特异性表达、识别顺式调控元件以及表征基因组变异,从而能够更精确地研究基因型与表型之间的关系。第三代单分子长读长和合成共条形码读长测序技术的最新进展利用了长程信息来简化组装图并改善基因组序列组装。然而,由于长读长的测序错误率高以及共条形码读长的捕获效率有限,重建完整的单倍型在方法上仍然具有挑战性。我们在此提出了一种名为AsmMix的流程,用于生成连续且准确的二倍体基因组。它首先组装共条形码读长以生成可能包含许多缺口的准确的单倍型解析组装,而长读长组装是连续的但容易出错。然后将两个组装集整合到错误组装减少的单倍型解析组装中。通过对多个合成数据集的广泛评估,AsmMix在各种测序平台、覆盖深度、读长和读长准确性上始终展示出用于单倍型分型的高精度和召回率,显著优于该领域的其他现有工具。此外,我们使用人类全基因组数据集(HG002)验证了我们流程的有效性,并生成了高度连续、准确且单倍型解析的组装。这些组装使用GIAB基准进行评估,证实了变异检测的准确性。我们的结果表明,AsmMix提供了一种直接而高效的方法,有效地利用长读长和共条形码读长进行单倍型解析组装。