Qi Yanwei, Gu Shengqiang, Zhang Yue, Guo Lidong, Xu Mengyang, Cheng Xiaofang, Wang Ou, Sun Ying, Chen Jianwei, Fang Xiaodong, Liu Xin, Deng Li, Fan Guangyi
BGI-Qingdao BGI-Shenzhen Qingdao China.
State Key Laboratory of Agricultural Genomics BGI-Shenzhen Shenzhen China.
Imeta. 2022 Aug 15;1(4):e46. doi: 10.1002/imt2.46. eCollection 2022 Dec.
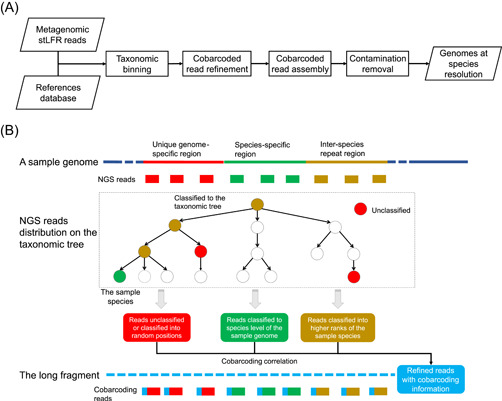
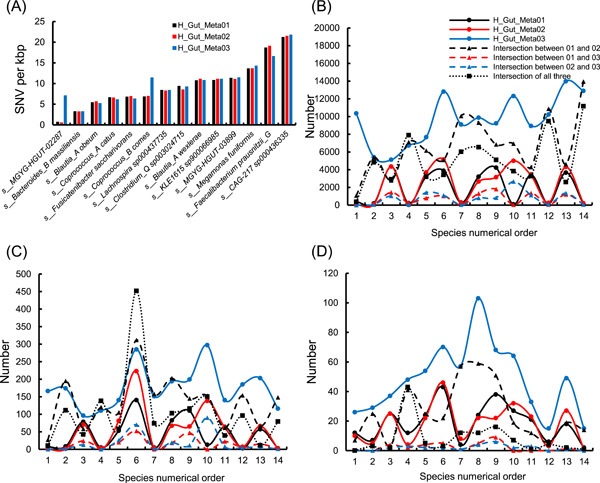
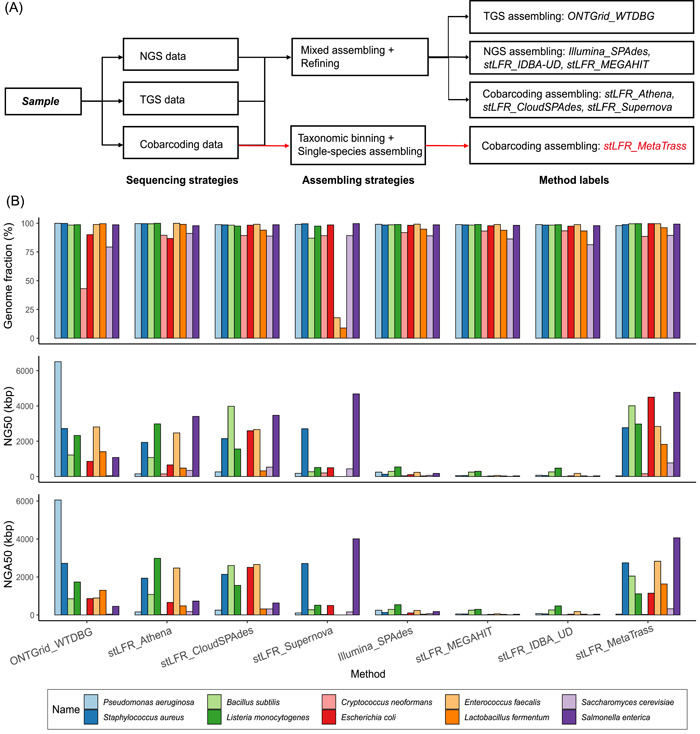
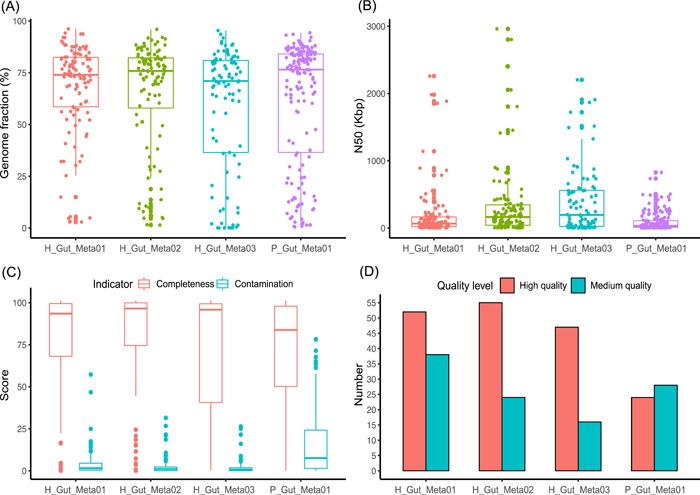
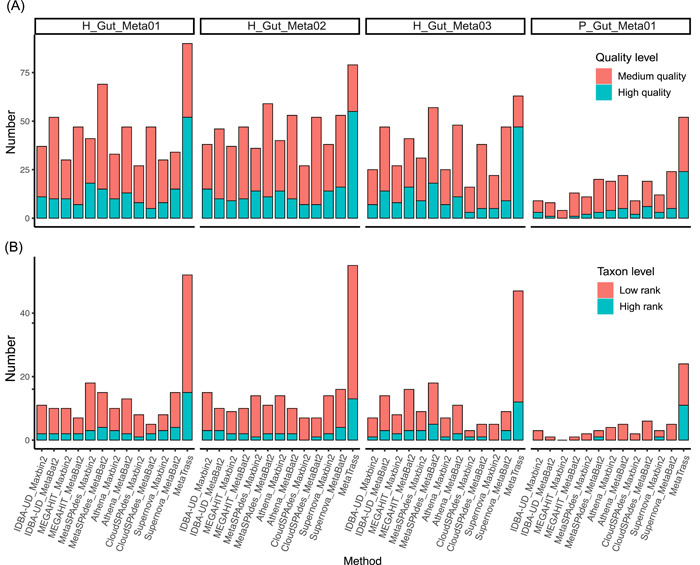
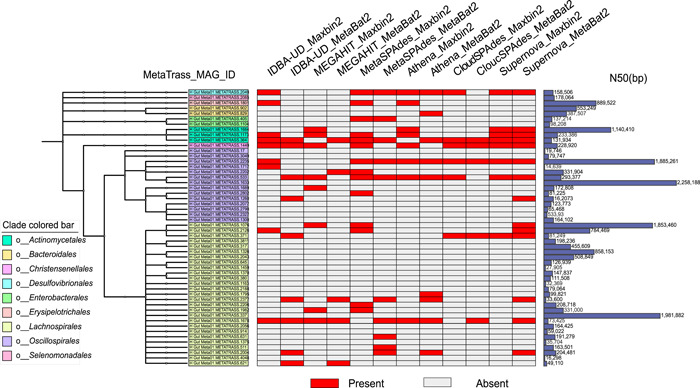
Metagenomic evidence of great genetic diversity within the nonconserved regions of the human gut microbial genomes appeals for new methods to elucidate the species-level variability at high resolution. However, current approaches cannot satisfy this methodologically challenge. In this study, we proposed an efficient binning-first-and-assembly-later strategy, named MetaTrass, to recover high-quality species-resolved genomes based on public reference genomes and the single-tube long fragment read (stLFR) technology, which enables cobarcoding. MetaTrass can generate genomes with longer contiguity, higher completeness, and lower contamination than those produced by conventional assembly-first-and-binning-later strategies. From a simulation study on a mock microbial community, MetaTrass showed the potential to improve the contiguity of assembly from kb to Mb without accuracy loss, as compared to other methods based on the next-generation sequencing technology. From four human fecal samples, MetaTrass successfully retrieved 178 high-quality genomes, whereas only 58 ones were provided by the optimal performance of other conventional strategies. Most importantly, these high-quality genomes confirmed the high level of genetic diversity among different samples and unveiled much more. MetaTrass was designed to work with metagenomic reads sequenced by stLFR technology, but is also applicable to other types of cobarcoding libraries. With the high capability of assembling high-quality genomes of metagenomic data sets, MetaTrass seeks to facilitate the study of spatial characters and dynamics of complex microbial communities at enhanced resolution. The open-source code of MetaTrass is available at https://github.com/BGI-Qingdao/MetaTrass.
人类肠道微生物基因组非保守区域内存在巨大遗传多样性的宏基因组学证据,促使人们寻求新方法以高分辨率阐明物种水平的变异性。然而,目前的方法无法应对这一方法学挑战。在本研究中,我们提出了一种高效的先分箱后组装策略,名为MetaTrass,以基于公共参考基因组和单管长片段读取(stLFR)技术(该技术支持共条形码编码)来恢复高质量的物种解析基因组。与传统的先组装后分箱策略相比,MetaTrass能够生成具有更长连续性、更高完整性和更低污染的基因组。通过对模拟微生物群落的研究,与基于下一代测序技术的其他方法相比,MetaTrass显示出在不损失准确性的情况下将组装连续性从kb提高到Mb的潜力。从四个人类粪便样本中,MetaTrass成功检索到178个高质量基因组,而其他传统策略的最佳性能仅提供了58个。最重要的是,这些高质量基因组证实了不同样本间高水平的遗传多样性,并揭示了更多信息。MetaTrass旨在与通过stLFR技术测序的宏基因组读取数据配合使用,但也适用于其他类型的共条形码文库。凭借其组装宏基因组数据集高质量基因组的强大能力,MetaTrass致力于在更高分辨率下促进对复杂微生物群落空间特征和动态的研究。MetaTrass的开源代码可在https://github.com/BGI-Qingdao/MetaTrass获取。