School of Molecular Sciences, Arizona State University, Tempe, AZ, USA.
Center for Innovations in Medicine, Biodesign Institute, Arizona State University, Tempe, AZ, USA.
Commun Biol. 2024 Aug 12;7(1):979. doi: 10.1038/s42003-024-06650-3.
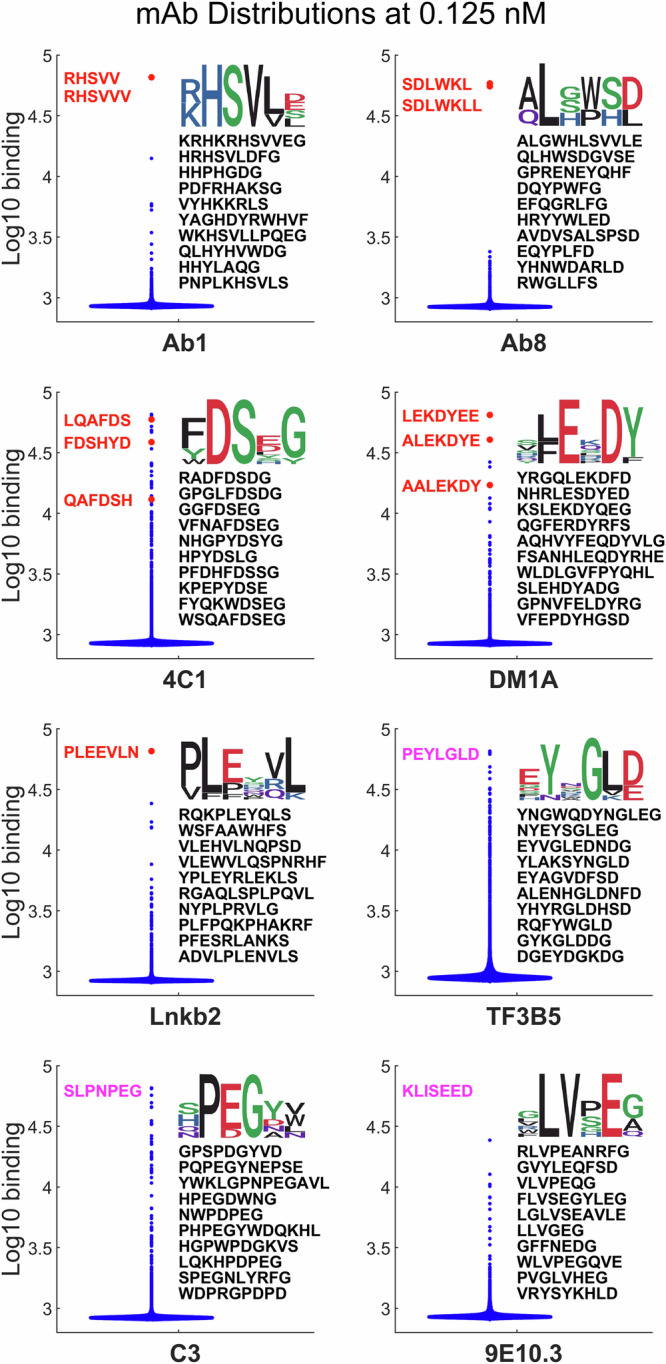
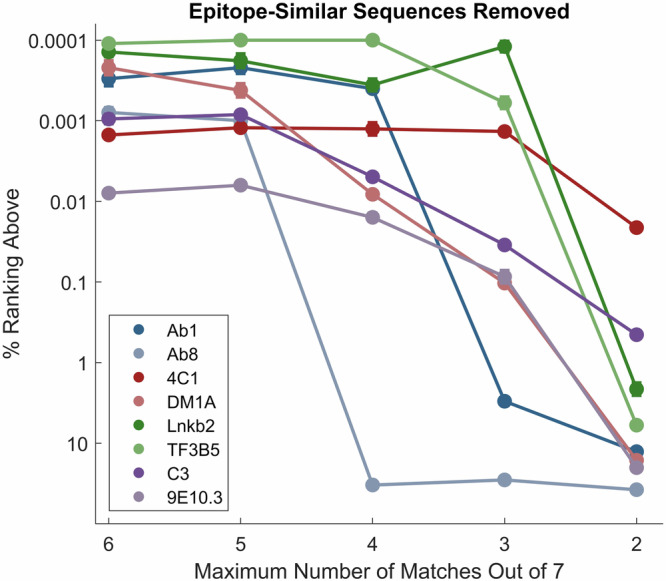
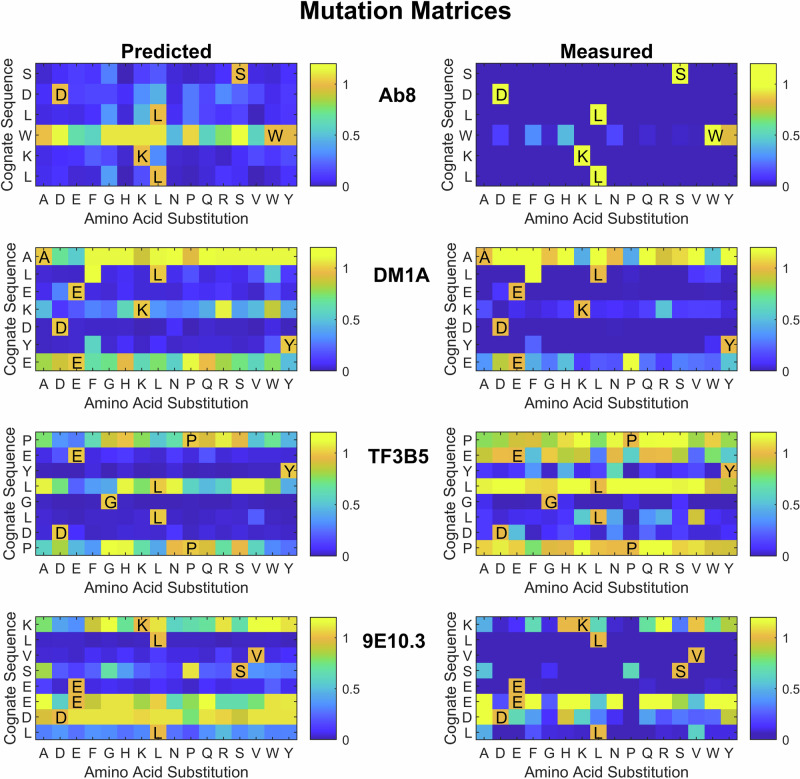
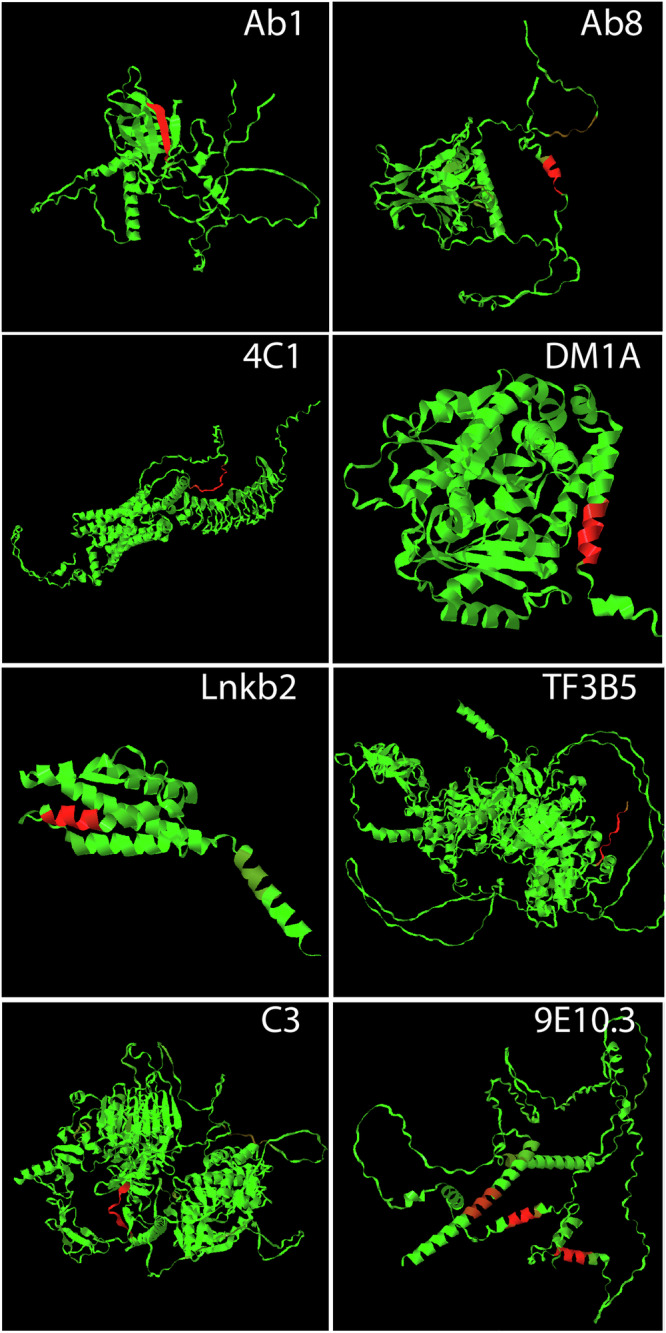
Previous work has shown that binding of target proteins to a sparse, unbiased sample of all possible peptide sequences is sufficient to train a machine learning model that can then predict, with statistically high accuracy, target binding to any possible peptide sequence of similar length. Here, highly sequence-specific molecular recognition is explored by measuring binding of 8 monoclonal antibodies (mAbs) with specific linear cognate epitopes to an array containing 121,715 near-random sequences about 10 residues in length. Network models trained on resulting sequence-binding values are used to predict the binding of each mAb to its cognate sequence and to an in silico generated one million random sequences. The model always ranks the binding of the cognate sequence in the top 100 sequences, and for 6 of the 8 mAbs, the cognate sequence ranks in the top ten. Practically, this approach has potential utility in selecting highly specific mAbs for therapeutics or diagnostics. More fundamentally, this demonstrates that very sparse random sampling of a large amino acid sequence spaces is sufficient to generate comprehensive models predictive of highly specific molecular recognition.
先前的工作表明,将目标蛋白与所有可能肽序列的稀疏、无偏样本结合足以训练机器学习模型,该模型可以以前所未有的统计精度预测任何类似长度的目标肽序列的结合。在这里,通过测量 8 种具有特定线性同源表位的单克隆抗体 (mAb) 与包含约 121,715 个长度为 10 个残基的近随机序列的阵列的结合,探索了高度序列特异性的分子识别。基于所得序列结合值训练的网络模型用于预测每个 mAb 与其同源序列和计算生成的 100 万个随机序列的结合。该模型始终将同源序列的结合排在前 100 个序列中,对于 8 种 mAb 中的 6 种,同源序列排在前 10 位。实际上,这种方法在选择用于治疗或诊断的高度特异性 mAb 方面具有潜在的用途。更根本的是,这表明对大氨基酸序列空间进行非常稀疏的随机采样足以生成可预测高度特异性分子识别的综合模型。