Al-Hussaini Abdulrahman, Alrashidi Sami, Hafez Deema H, Alkhalifah Yasir S, Otayn Bashaer, Alrasheed Majid, Al Mufarreh Sumayah, AlKasim Sultan
Division of Pediatric Gastroenterology, Children's Specialized Hospital, King Fahad Medical City, Riyadh, Saudi Arabia.
College of Medicine, Alfaisal University, Riyadh, Saudi Arabia.
Front Pediatr. 2024 Jul 30;12:1423657. doi: 10.3389/fped.2024.1423657. eCollection 2024.
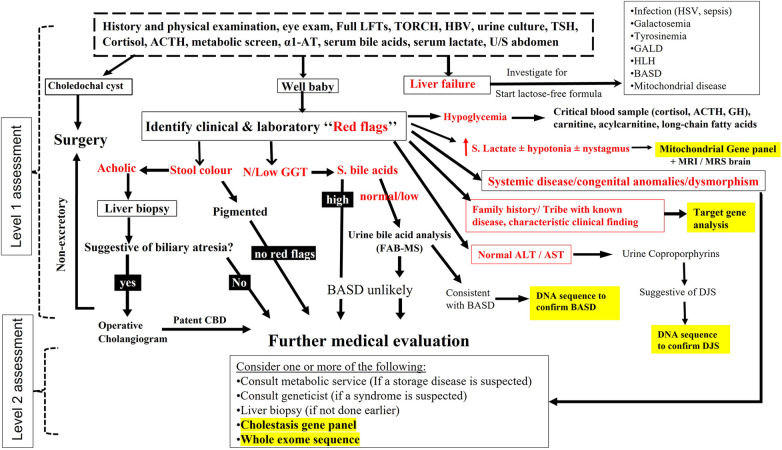
Most of the literature on infantile cholestasis (IC) originated from Caucasian and Asian populations. The differential diagnosis of IC is very broad, and identification of etiology is challenging to clinicians because the list includes many entities with overlapping clinical, biochemical, and histological features. Thus, a structured, stepwise diagnostic approach is required to help early recognition and prompt evaluation and management of treatable causes of cholestasis.
(1) To determine the differential diagnosis of IC among Saudi population and (2) to evaluate the usefulness of a diagnostic algorithm that has been tailored by the authors to the local practice.
All infants with onset of cholestasis before 12 months of age (2007 and 2020) were identified and included if they underwent extensive work up to exclude infectious, structural, metabolic, endocrine, infiltrative, and familial causes.
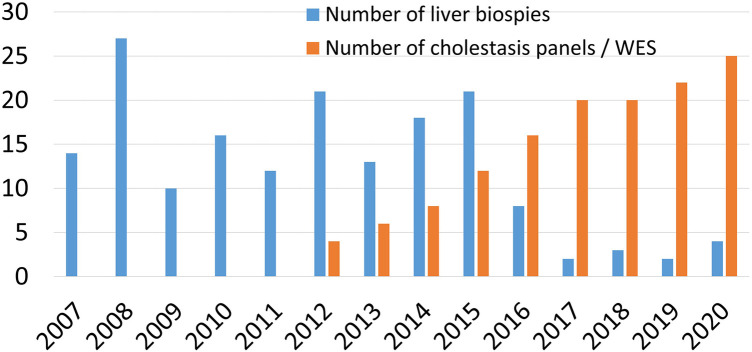
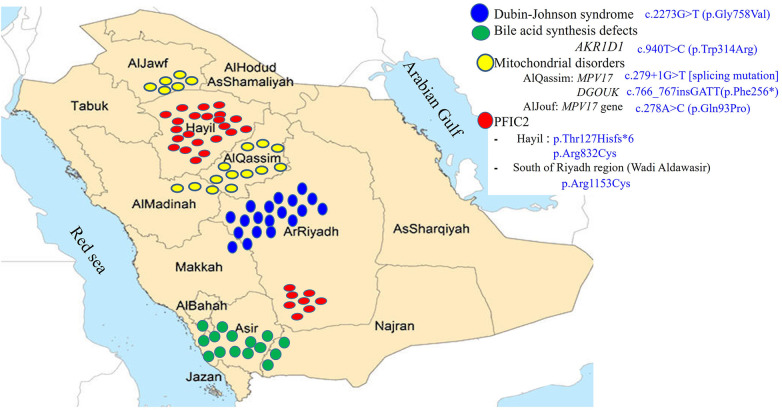
Our diagnostic pathway allowed a definite diagnosis in 373 of the included 533 cases; 160 (30%) were labelled as "idiopathic neonatal hepatitis" (INH) [i.e., overall 70% detection rate]. However, when considering the cases that underwent extensive investigations including advanced gene testing (415 of the 533), the yield of the diagnostic algorithm was 90% (373/415). Familial cholestasis group was the most common in 20% (107/533), and biliary atresia and neonatal-onset Dubin Johnson syndrome contributed to 6% each. The genetic/hereditary causes of cholestasis contributed to 58% of the diagnosed cases (217/373). No single case of alpha-1 antitrypsin deficiency was diagnosed. Forty-nine infants with cholestasis presented with liver failure (9%).
Our study highlights several unique features and causes of IC among Arabs which could have a great impact on the differential diagnosis process and the choice of laboratory tests used in the clinical setting.
大多数关于婴儿胆汁淤积症(IC)的文献来自白种人和亚洲人群。IC的鉴别诊断范围非常广泛,病因识别对临床医生来说具有挑战性,因为其中包括许多具有重叠临床、生化和组织学特征的疾病。因此,需要一种结构化、逐步的诊断方法来帮助早期识别并及时评估和处理胆汁淤积症的可治疗病因。
(1)确定沙特人群中IC的鉴别诊断;(2)评估作者根据当地实际情况定制的诊断算法的实用性。
确定所有在12个月龄前出现胆汁淤积症(2007年至2020年)的婴儿,若他们接受了广泛检查以排除感染、结构、代谢、内分泌、浸润性和家族性病因,则纳入研究。
我们的诊断途径在纳入的533例病例中的373例中做出了明确诊断;160例(30%)被标记为“特发性新生儿肝炎”(INH)[即总体检出率为70%]。然而,当考虑接受了包括先进基因检测在内的广泛检查的病例(533例中的415例)时,诊断算法的阳性率为90%(373/415)。家族性胆汁淤积症组最为常见,占20%(107/533),胆道闭锁和新生儿期发病的杜宾-约翰逊综合征各占6%。胆汁淤积症的遗传/遗传性病因占确诊病例的58%(217/373)。未诊断出一例α-1抗胰蛋白酶缺乏症。49例胆汁淤积症婴儿出现肝功能衰竭(9%)。
我们的研究突出了阿拉伯人群中IC的几个独特特征和病因,这可能对鉴别诊断过程以及临床环境中使用的实验室检查的选择产生重大影响。