Gupta Ajay Kumar, Vaishnav Yogesh, Jain Sanmati Kumar, Annadurai Sivakumar, Kumar Neeraj
Drug Discovery and Research Laboratory, Department of Pharmacy, Guru Ghasidas Vishwavidyalaya (A Central University), Bilaspur, Chhattisgarh, India.
Department of Pharmacognosy, College of Pharmacy, King Khalid University, Abha, Saudi Arabia.
Front Chem. 2024 Aug 6;12:1418975. doi: 10.3389/fchem.2024.1418975. eCollection 2024.
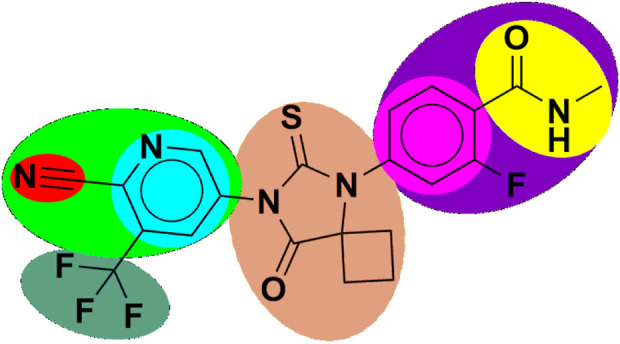
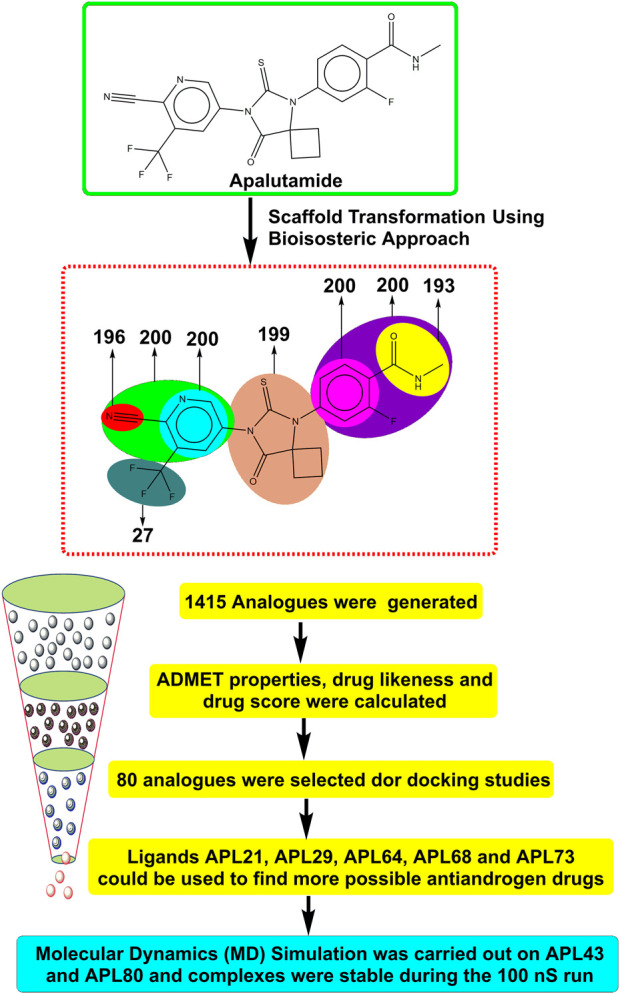
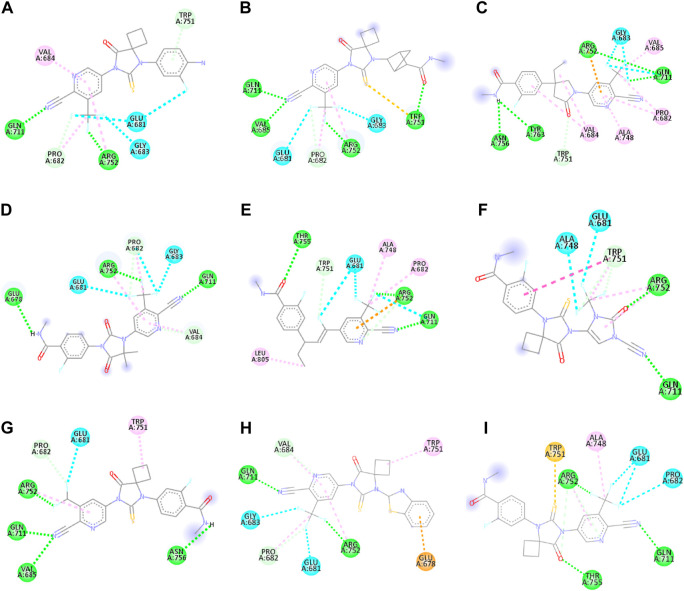
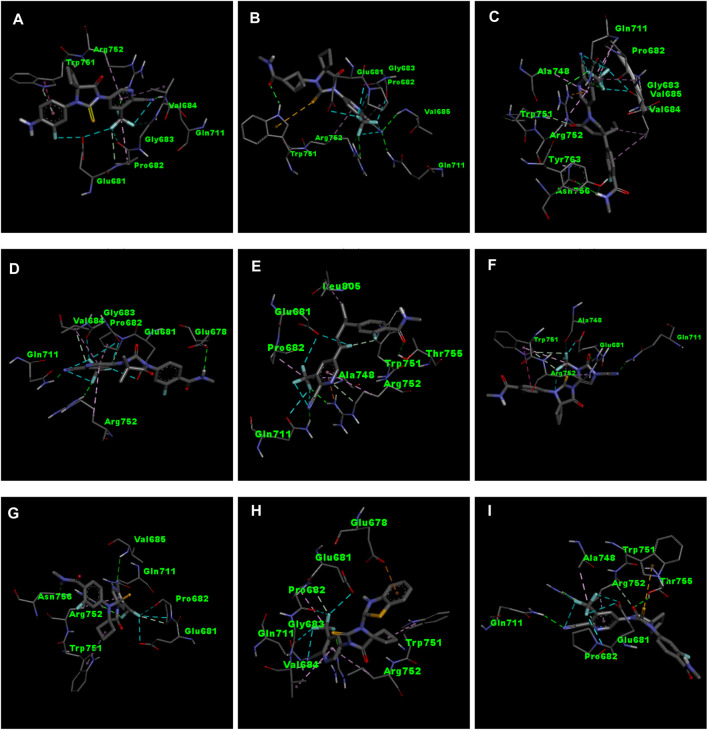
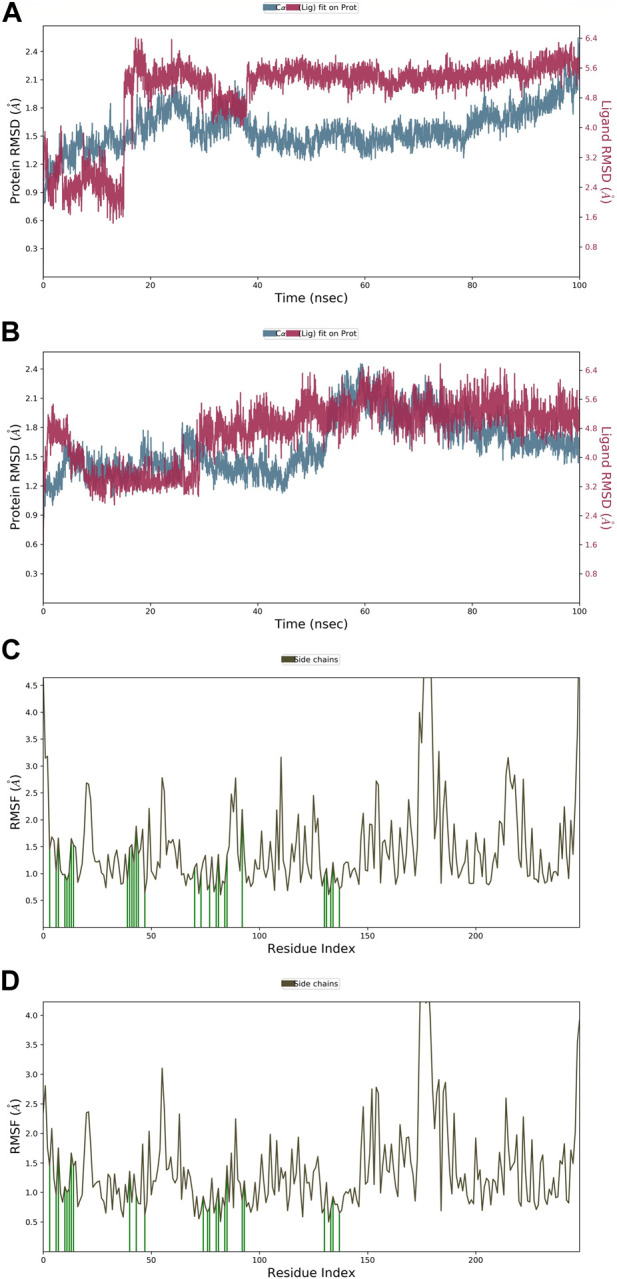
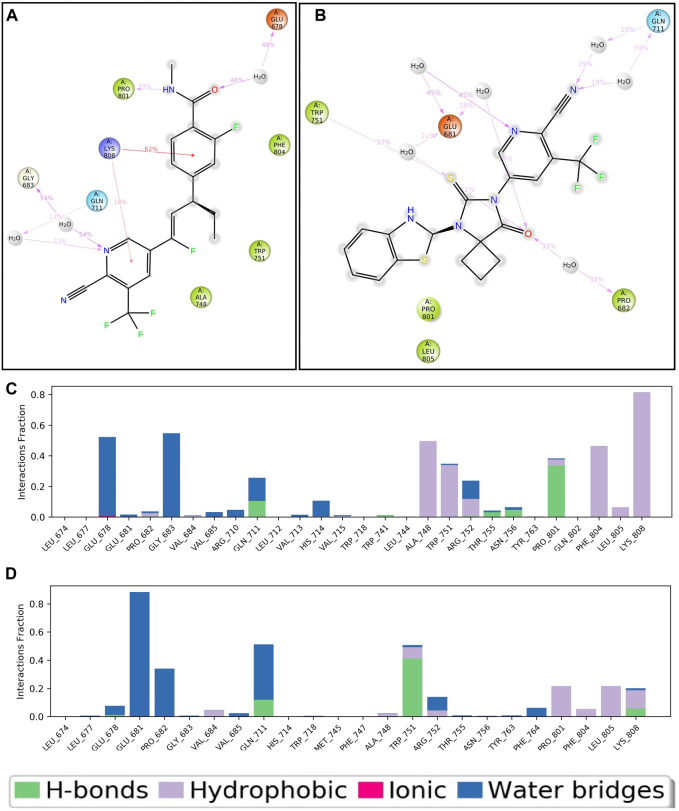
Prostate cancer (PC) ranks as the second most frequent type of cancer in men and is the fourth largest cause of mortality worldwide. Androgenic hormones such as testosterone and dihydrotestosterone are crucial for the development and progression of the prostate gland. Androgenic hormones bind to androgen receptors (AR) and trigger the synthesis of many genes that stimulate the growth of prostate cells, initiating PC growth. Apalutamide (APL) is a non-steroidal antiandrogen drug used to treat PC; however, it also causes a variety of toxicities and resistance during the treatment. The purpose of this study was to computationally identify new and safer analogues of APL, focusing on improved pharmacokinetic properties and reduced toxicity. Drug likeness (DL) and drug score (DS) were also calculated. Docking studies on the designed analogues were conducted to predict their binding affinities and compare their orientations with the ligands in the original crystal structure. Molecular dynamics (MD) simulation of docked ligands was done using Schrödinger suite. We generated a total of 1,415 analogues for different groups of APL using the bioisosteric approach. We selected 80 bioisosteres based on pharmacokinetic profiles, DL and DS score predictions, and found that the designed APL bioisosteres were optimal to good compared to APL. Analogues APL19, APL35, APL43, APL76, and APL80, formed hydrogen bonds with protein () which is similar hydrogen bonding to the standard (APL). The MD simulation result confirmed that APL43 and APL80 complexes were stable during the 100 nS run. The results suggest that the APL analogues, particularly APL43 and APL80, are predicted to be potential antiandrogen drugs for the treatment of prostate cancer.
前列腺癌(PC)是男性中第二常见的癌症类型,也是全球第四大死因。睾酮和双氢睾酮等雄激素对前列腺的发育和进展至关重要。雄激素与雄激素受体(AR)结合,触发许多刺激前列腺细胞生长的基因的合成,从而引发前列腺癌的生长。阿帕鲁胺(APL)是一种用于治疗前列腺癌的非甾体抗雄激素药物;然而,它在治疗过程中也会引起各种毒性和耐药性。本研究的目的是通过计算识别APL的新型且更安全的类似物,重点是改善药代动力学性质并降低毒性。还计算了药物相似性(DL)和药物评分(DS)。对设计的类似物进行对接研究,以预测它们的结合亲和力,并将它们的取向与原始晶体结构中的配体进行比较。使用薛定谔套件对对接的配体进行分子动力学(MD)模拟。我们使用生物电子等排体方法为不同组的APL总共生成了1415个类似物。我们根据药代动力学概况、DL和DS评分预测选择了80个生物电子等排体,发现与APL相比,设计的APL生物电子等排体为最优至良好。类似物APL19、APL35、APL43、APL76和APL80与蛋白质()形成氢键,这与标准物(APL)的氢键相似。MD模拟结果证实,APL43和APL80复合物在100纳秒的运行过程中是稳定的。结果表明,APL类似物,特别是APL43和APL80,预计是治疗前列腺癌的潜在抗雄激素药物。