Independent Researcher, Melbourne, VIC 3000, Australia.
Faculty of Science, University of Melbourne, Parkville, VIC 3010, Australia.
Int J Mol Sci. 2024 Aug 7;25(16):8614. doi: 10.3390/ijms25168614.
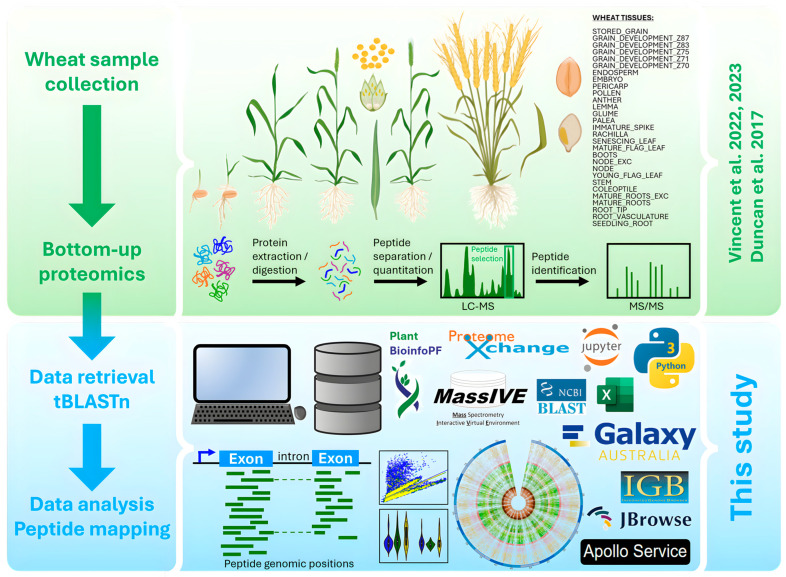
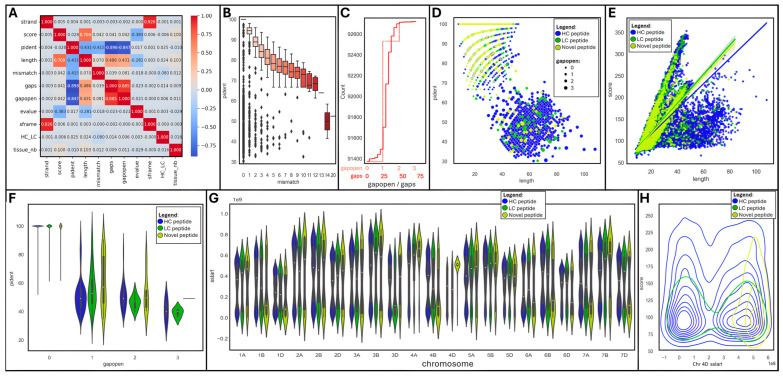
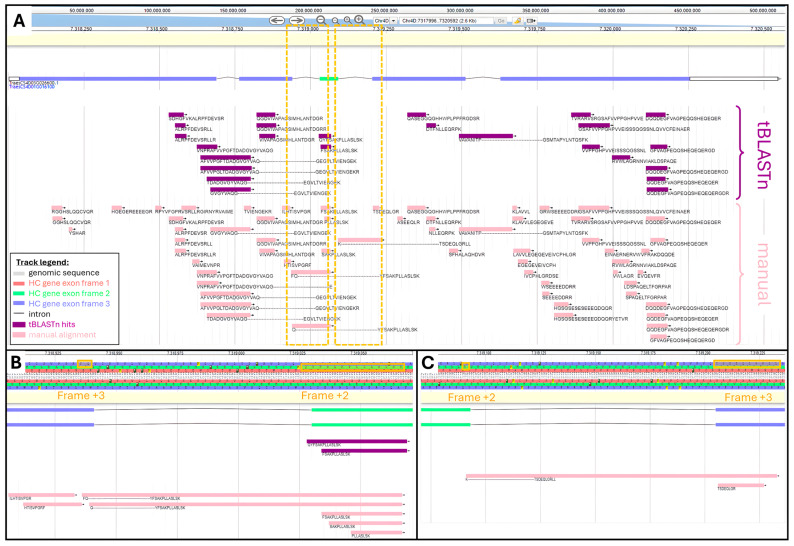
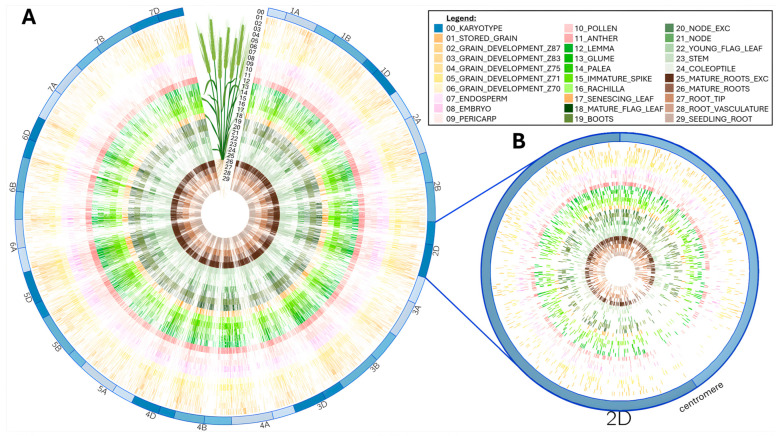
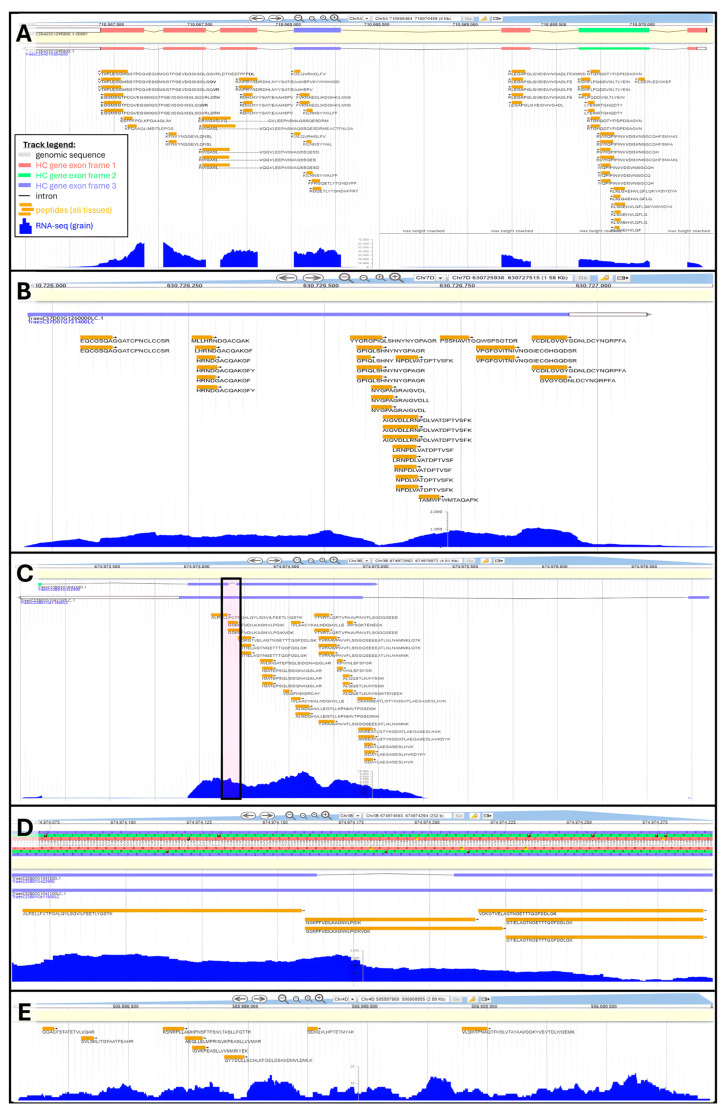
is an important crop whose reference genome (International Wheat Genome Sequencing Consortium (IWGSC) RefSeq v2.1) offers a valuable resource for understanding wheat genetic structure, improving agronomic traits, and developing new cultivars. A key aspect of gene model annotation is protein-level evidence of gene expression obtained from proteomics studies, followed up by proteogenomics to physically map proteins to the genome. In this research, we have retrieved the largest recent wheat proteomics datasets publicly available and applied the Basic Local Alignment Search Tool (tBLASTn) algorithm to map the 861,759 identified unique peptides against IWGSC RefSeq v2.1. Of the 92,719 hits, 83,015 unique peptides aligned along 33,612 High Confidence (HC) genes, thus validating 31.4% of all wheat HC gene models. Furthermore, 6685 unique peptides were mapped against 3702 Low Confidence (LC) gene models, and we argue that these gene models should be considered for HC status. The remaining 2934 orphan peptides can be used for novel gene discovery, as exemplified here on chromosome 4D. We demonstrated that tBLASTn could not map peptides exhibiting mid-sequence frame shift. We supply all our proteogenomics results, Galaxy workflow and Python code, as well as Browser Extensible Data (BED) files as a resource for the wheat community via the Apollo Jbrowse, and GitHub repositories. Our workflow could be applied to other proteomics datasets to expand this resource with proteins and peptides from biotically and abiotically stressed samples. This would help tease out wheat gene expression under various environmental conditions, both spatially and temporally.
是一种重要的作物,其参考基因组(国际小麦基因组测序联盟(IWGSC)RefSeq v2.1)为了解小麦遗传结构、改良农艺性状和培育新品种提供了宝贵的资源。基因模型注释的一个关键方面是从蛋白质组学研究中获得的蛋白质水平的基因表达证据,随后通过蛋白质基因组学将蛋白质物理映射到基因组上。在这项研究中,我们检索了最近公开的最大的小麦蛋白质组学数据集,并应用基本局部比对搜索工具(tBLASTn)算法将 861,759 个鉴定的独特肽映射到 IWGSC RefSeq v2.1。在 92,719 个命中中,83,015 个独特肽沿着 33,612 个高可信度(HC)基因对齐,从而验证了所有小麦 HC 基因模型的 31.4%。此外,6685 个独特肽被映射到 3702 个低可信度(LC)基因模型上,我们认为这些基因模型应该被考虑为 HC 状态。其余的 2934 个孤儿肽可用于新基因的发现,这里在 4D 染色体上举例说明了这一点。我们证明 tBLASTn 无法映射具有中间序列移码的肽。我们通过 Apollo Jbrowse 和 GitHub 存储库,为小麦社区提供了所有的蛋白质基因组学结果、Galaxy 工作流程和 Python 代码,以及 Browser Extensible Data(BED)文件。我们的工作流程可以应用于其他蛋白质组学数据集,以从生物和非生物胁迫的样本中扩展该资源的蛋白质和肽。这将有助于梳理小麦在各种环境条件下的基因表达,包括空间和时间上的表达。