Epilepsy Research Group, Clinical and Health Sciences, Australian Centre for Precision Health, University of South Australia, Adelaide, SA 5000, Australia.
Novocraft Technologies, Petaling Jaya 46300, Malaysia.
Genes (Basel). 2024 Aug 5;15(8):1031. doi: 10.3390/genes15081031.
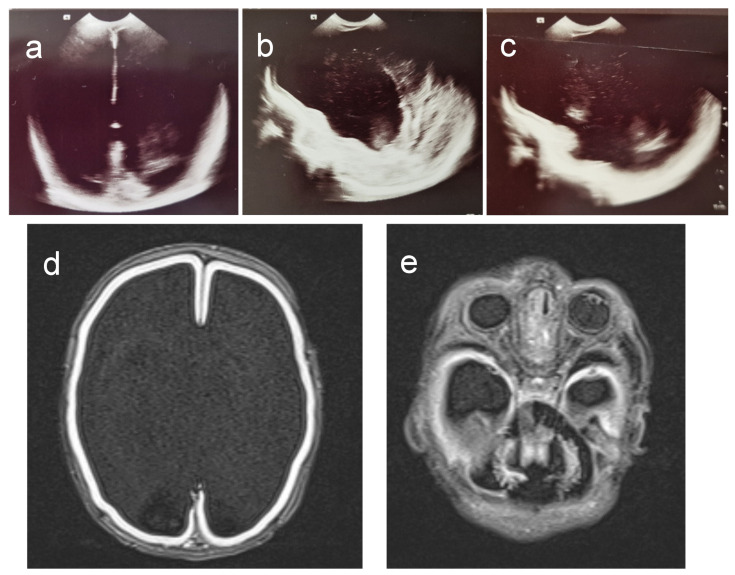
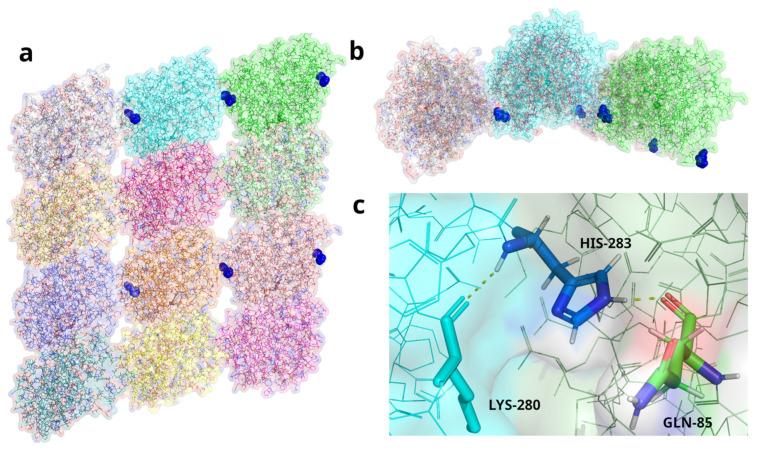
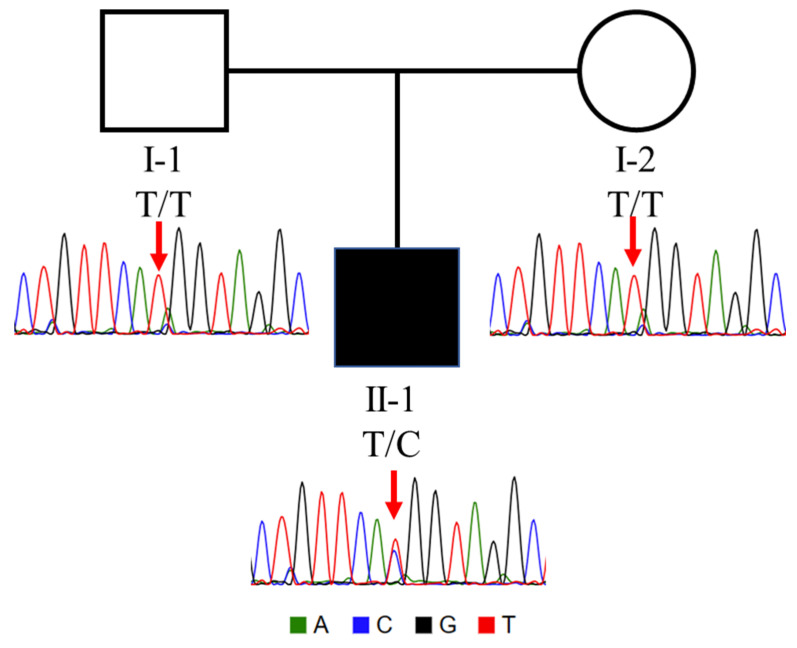
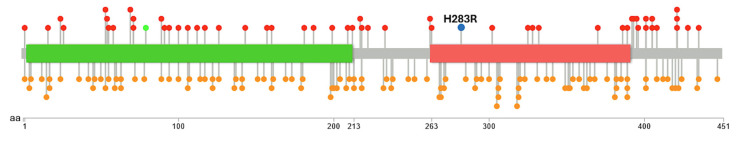
Tubulinopathies are associated with malformations of cortical development but not Walker-Warburg Syndrome. Intensive monitoring of a Croatian infant presenting as Walker-Warburg Syndrome in utero began at 21 weeks due to increased growth of cerebral ventricles and foetal biparietal diameter. Monitoring continued until Caesarean delivery at 34 weeks where the infant was eutrophic. Clinical assessment of a progressive neurological disorder of unknown aetiology found a macrocephalic head and markedly hypoplastic genitalia with a micropenis. Neurological examination showed generalized hypotonia with very rare spontaneous movements, hypotonia-induced respiratory insufficiency and ventilator dependence, and generalized myoclonus intensifying during manipulation. With clinical features of hypotonia, lissencephaly, and brain malformations, Walker-Warburg Syndrome was suspected; however, eye anomalies were absent. Genetic trio analysis via whole-exome sequencing only identified a novel de novo mutation in the gene (NM_006009.4:c.848A>G; NP_006000.2:p.His283Arg) in the infant, who died at 2 months of age, as the likely cause. We report a previously unpublished, very rare heterozygous mutation with clinical features of macrocephaly and hypoplastic genitalia which have not previously been associated with the gene. The absence of eye phenotypes or mutations in Walker-Warburg-associated genes confirm this as not a new presentation of Walker-Warburg Syndrome but a novel tubulinopathy for neonatologists to be aware of.
微管病与皮质发育畸形有关,但与沃克-沃伯格综合征无关。一名克罗地亚婴儿在宫内表现为沃克-沃伯格综合征,由于脑室和胎儿双额径的生长增加,从 21 周开始进行密集监测。监测一直持续到 34 周进行剖宫产,此时婴儿为正营养状态。对病因不明的进行性神经功能障碍进行临床评估,发现头颅巨大,生殖器明显发育不良,伴有小阴茎。神经学检查显示全身性低张力,自发运动非常罕见,低张力引起的呼吸功能不全和对呼吸机的依赖,以及在操作过程中全身性肌阵挛加剧。由于低张力、无脑回和脑畸形的临床特征,怀疑为沃克-沃伯格综合征;然而,没有眼部异常。通过全外显子组测序进行的遗传三联体分析仅在婴儿中发现了一个新的从头突变(NM_006009.4:c.848A>G;NP_006000.2:p.His283Arg),该婴儿在 2 个月大时死亡,这可能是导致这种疾病的原因。我们报告了一个以前未发表的、非常罕见的杂合性 突变,具有大头畸形和生殖器发育不良的临床特征,这些特征以前与该基因无关。没有眼部表型或沃克-沃伯格相关基因的突变证实这不是沃克-沃伯格综合征的新表现,而是一种新的 微管病,新生儿科医生需要注意。