Cancer Center and Research Institute, University of Mississippi Medical Center, Jackson, Mississippi, USA; Department of Cell and Molecular Biology, University of Mississippi Medical Center, Jackson, Mississippi, USA.
Mitchell Cancer Institute, University of South Alabama, Mobile, Alabama, USA; Department of Pathology, University of South Alabama, Mobile, Alabama, USA.
J Biol Chem. 2024 Oct;300(10):107753. doi: 10.1016/j.jbc.2024.107753. Epub 2024 Sep 10.
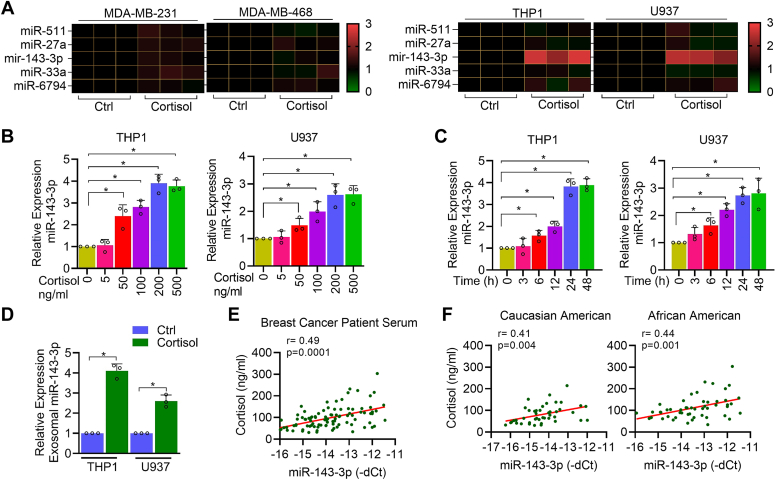
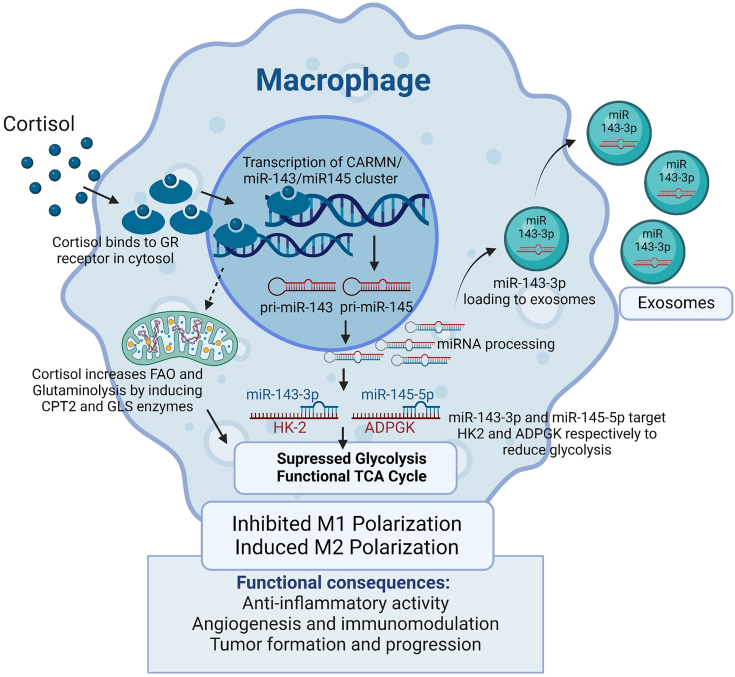
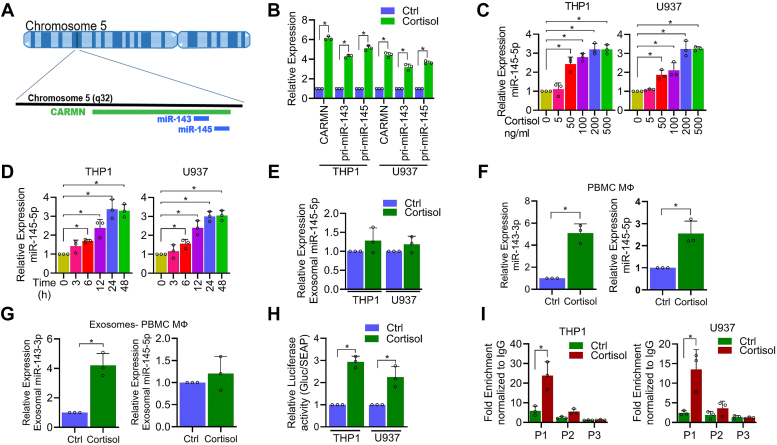
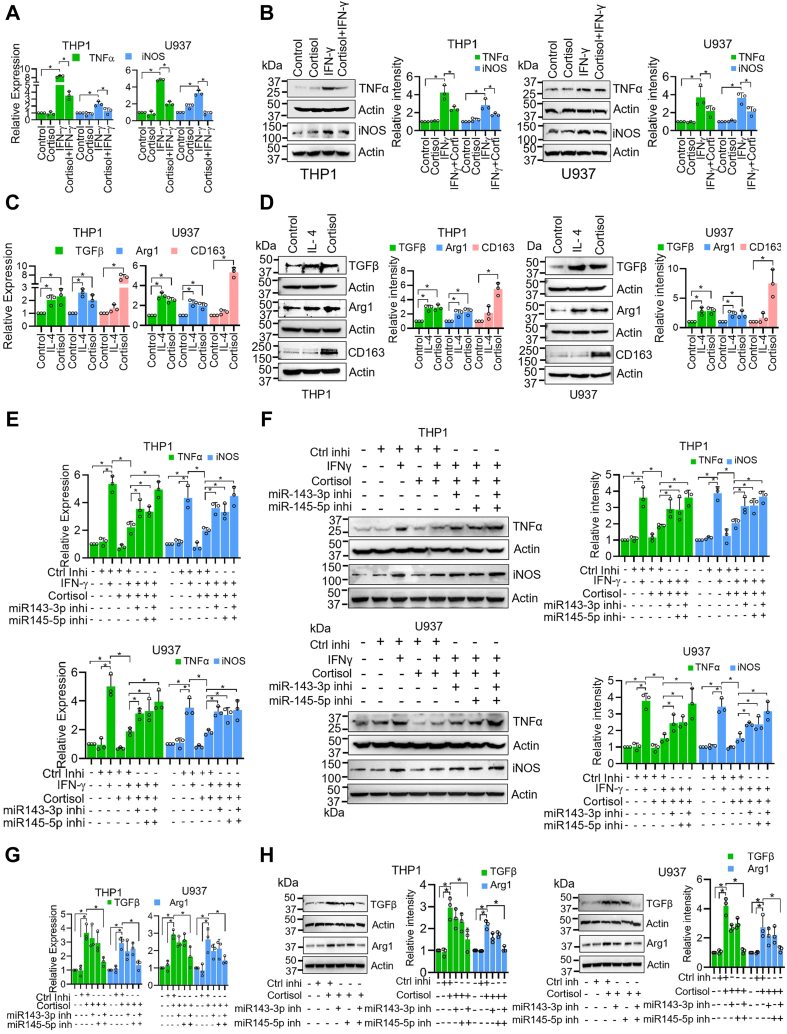
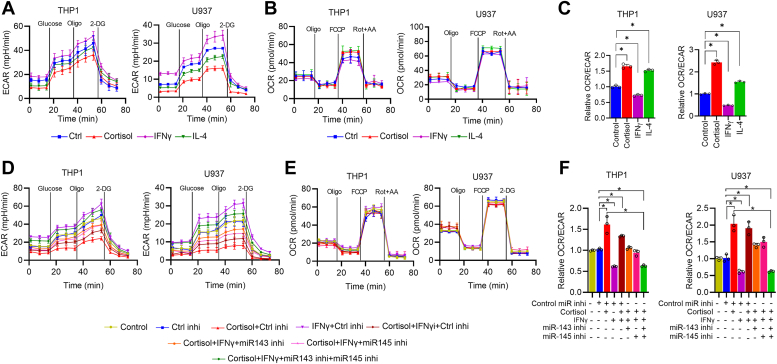
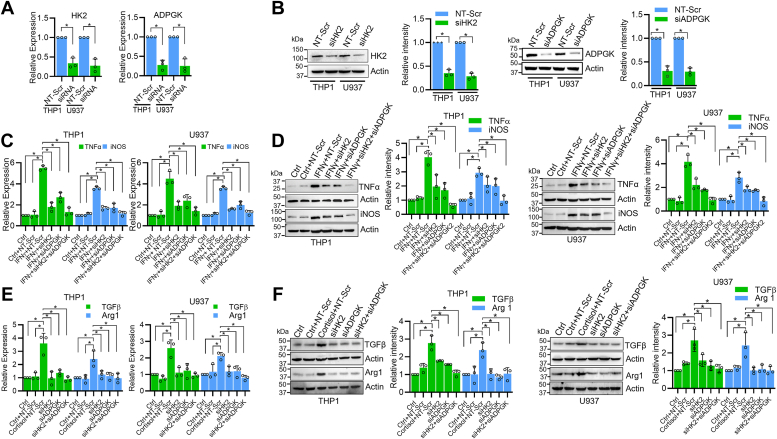
Chronic stress can have adverse consequences on human health by disrupting the hormonal balance in our body. Earlier, we observed elevated levels of cortisol, a primary stress hormone, and some exosomal microRNAs in the serum of patients with breast cancer. Here, we investigated the role of cortisol in microRNA induction and its functional consequences. We found that cortisol induced the expression of miR-143/145 cluster in human monocyte (THP1 and U937)-derived macrophages but not in breast cancer cells. In silico analysis identified glucocorticoid-response element in the upstream CARMN promoter utilized by the miR-143/145 cluster. Enhanced binding of glucocorticoid-receptor (GR) upon cortisol exposure and its regulatory significance was confirmed by chromatin-immunoprecipitation and promoter-reporter assays. Further, cortisol inhibited IFNγ-induced M1 polarization and promoted M2 polarization, and these effects were suppressed by miR-143-3p and miR-145-5p inhibitors pretreatment. Cortisol-treated macrophages exhibited increased oxygen-consumption rate (OCR) to extracellular-acidification rate (ECAR) ratio, and this change was neutralized by functional inhibition of miR-143-3p and miR-145-5p. HK2 and ADPGK were confirmed as the direct targets of miR-143-3p and miR-145-5p, respectively. Interestingly, silencing of HK2 and ADPGK inhibited IFNγ-induced M1 polarization but failed to induce M2 polarization, since it suppressed both ECAR and OCR, while OCR was largely sustained in cortisol-treated M2-polarized macrophages. We found that cortisol treatment sustained OCR by enhancing fatty acid and glutamine metabolism through upregulation of CPT2 and GLS, respectively, to support M2 polarization. Thus, our findings unfold a novel mechanism of immune suppression by cortisol and open avenues for preventive and therapeutic interventions.
慢性应激通过扰乱体内激素平衡对人类健康产生不良影响。我们之前观察到乳腺癌患者血清中皮质醇(一种主要的应激激素)和一些外泌体 microRNA 的水平升高。在这里,我们研究了皮质醇在 microRNA 诱导中的作用及其功能后果。我们发现皮质醇诱导人单核细胞(THP1 和 U937)衍生的巨噬细胞中 miR-143/145 簇的表达,但不在乳腺癌细胞中诱导。计算分析确定了 miR-143/145 簇利用的上游 CARMN 启动子中的糖皮质激素反应元件。在皮质醇暴露时,糖皮质激素受体 (GR) 的结合增强及其调控意义通过染色质免疫沉淀和启动子报告基因测定得到证实。此外,皮质醇抑制 IFNγ 诱导的 M1 极化并促进 M2 极化,而 miR-143-3p 和 miR-145-5p 抑制剂预处理抑制了这些作用。皮质醇处理的巨噬细胞表现出增加的耗氧率 (OCR) 与细胞外酸化率 (ECAR) 的比值,而 miR-143-3p 和 miR-145-5p 的功能抑制中和了这种变化。HK2 和 ADPGK 被确认为 miR-143-3p 和 miR-145-5p 的直接靶标。有趣的是,HK2 和 ADPGK 的沉默抑制了 IFNγ 诱导的 M1 极化,但未能诱导 M2 极化,因为它抑制了 ECAR 和 OCR,而在皮质醇处理的 M2 极化巨噬细胞中 OCR 则得到很大维持。我们发现,皮质醇通过分别上调 CPT2 和 GLS 来增强脂肪酸和谷氨酰胺代谢,从而维持 OCR,以支持 M2 极化。因此,我们的研究结果揭示了皮质醇免疫抑制的新机制,并为预防和治疗干预开辟了途径。