Paul. G. Allen School of Computer Science and Engineering, University of Washington, Seattle, WA, USA.
Department of Biotechnology, Graduate School of Engineering, Osaka University, Suita, Japan.
Nature. 2024 Sep;633(8030):662-669. doi: 10.1038/s41586-024-07935-7. Epub 2024 Sep 11.
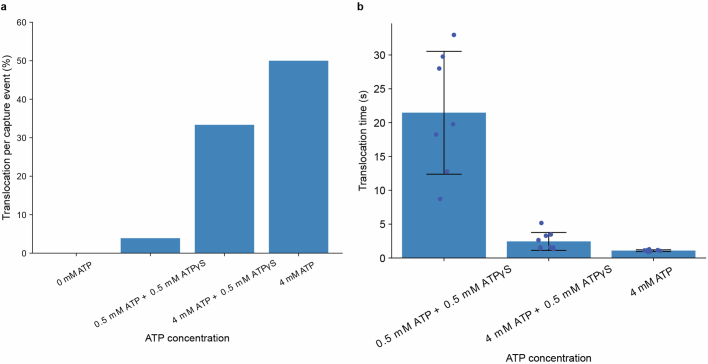
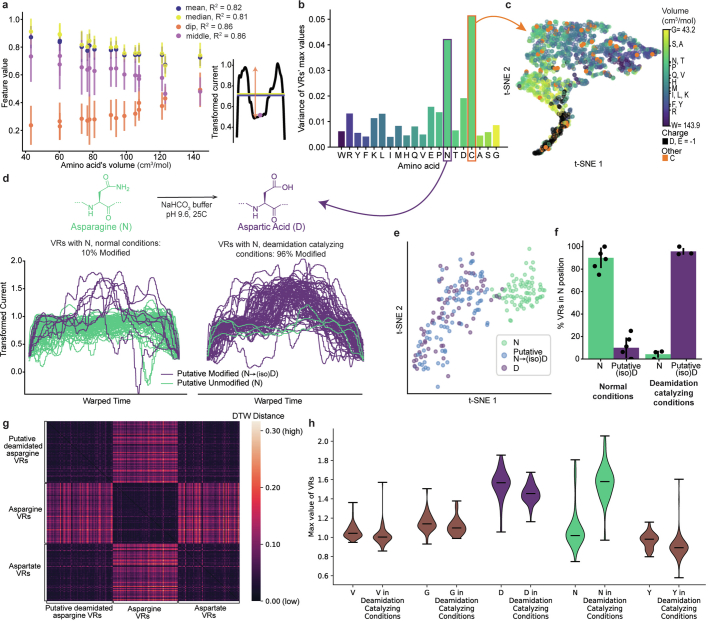
The ability to sequence single protein molecules in their native, full-length form would enable a more comprehensive understanding of proteomic diversity. Current technologies, however, are limited in achieving this goal. Here, we establish a method for the long-range, single-molecule reading of intact protein strands on a commercial nanopore sensor array. By using the ClpX unfoldase to ratchet proteins through a CsgG nanopore, we provide single-molecule evidence that ClpX translocates substrates in two-residue steps. This mechanism achieves sensitivity to single amino acids on synthetic protein strands hundreds of amino acids in length, enabling the sequencing of combinations of single-amino-acid substitutions and the mapping of post-translational modifications, such as phosphorylation. To enhance classification accuracy further, we demonstrate the ability to reread individual protein molecules multiple times, and we explore the potential for highly accurate protein barcode sequencing. Furthermore, we develop a biophysical model that can simulate raw nanopore signals a priori on the basis of residue volume and charge, enhancing the interpretation of raw signal data. Finally, we apply these methods to examine full-length, folded protein domains for complete end-to-end analysis. These results provide proof of concept for a platform that has the potential to identify and characterize full-length proteoforms at single-molecule resolution.
能够以其天然全长形式对单个蛋白质分子进行测序,将使我们能够更全面地了解蛋白质组的多样性。然而,目前的技术在实现这一目标方面存在局限性。在这里,我们在商业纳米孔传感器阵列上建立了一种用于长程、单分子读取完整蛋白质链的方法。通过使用 ClpX 解旋酶将蛋白质通过 CsgG 纳米孔棘轮化,我们提供了单分子证据,证明 ClpX 以两个残基的步骤转运底物。这种机制在数百个氨基酸长的合成蛋白质链上实现了对单个氨基酸的敏感性,从而能够对单个氨基酸取代的组合进行测序,并对翻译后修饰(如磷酸化)进行作图。为了进一步提高分类准确性,我们展示了多次重读数个单个蛋白质分子的能力,并探索了高度准确的蛋白质条形码测序的潜力。此外,我们开发了一种生物物理模型,该模型可以根据残基体积和电荷预先模拟原始纳米孔信号,从而增强对原始信号数据的解释。最后,我们应用这些方法来检查全长折叠蛋白质结构域,以进行完整的端到端分析。这些结果为一个平台提供了概念验证,该平台有可能以单分子分辨率识别和表征全长蛋白质异构体。