Yeow Dennis, Rudaks Laura Ivete, Davis Ryan, Ng Karl, Ghaoui Roula, Cheong Pak Leng, Ravenscroft Gianina, Kennerson Marina, Deveson Ira, Kumar Kishore Raj
Neurology Department and Molecular Medicine Laboratory, Concord Repatriation General Hospital, Sydney, New South Wales, Australia.
The University of Sydney Faculty of Medicine and Health, Sydney, New South Wales, Australia.
BMJ Neurol Open. 2025 May 11;7(1):e000990. doi: 10.1136/bmjno-2024-000990. eCollection 2025.
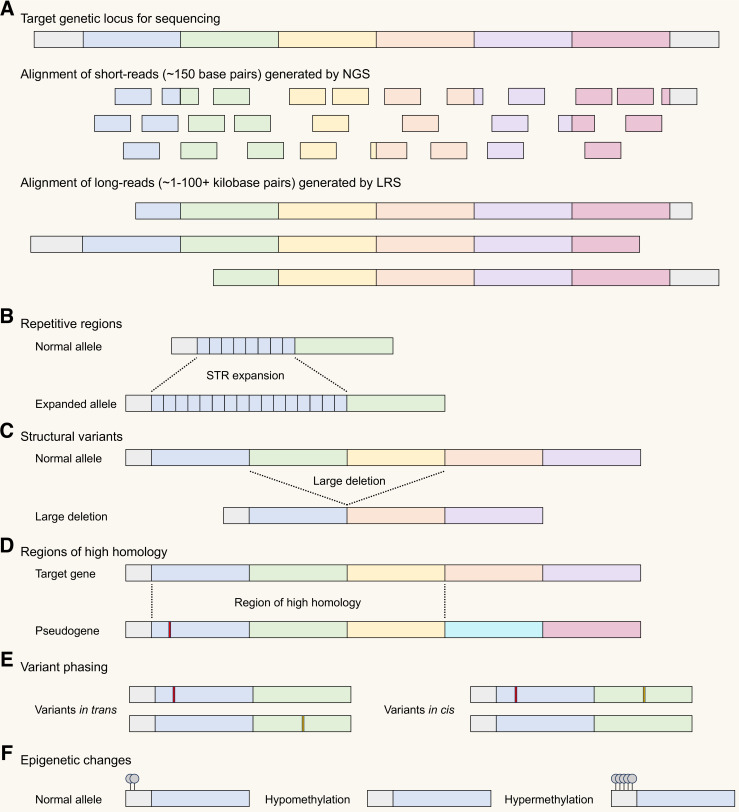
Genetic myopathies are caused by pathogenic variants in >300 genes across the nuclear and mitochondrial genomes. Although short-read next-generation sequencing (NGS) has revolutionised the diagnosis of genetic disorders, large and/or complex genetic variants, which are over-represented in the genetic myopathies, are not well characterised using this approach. Long-read sequencing (LRS) is a newer genetic testing technology that overcomes many of the limitations of NGS. In particular, LRS provides improved detection of challenging variant types, including short tandem repeat (STR) expansions, copy number variants and structural variants, as well as improved variant phasing and concurrent assessment of epigenetic changes, including DNA methylation. The ability to concurrently detect multiple STR expansions is particularly relevant given the growing number of recently described genetic myopathies associated with STR expansions. LRS will also aid in the identification of new myopathy genes and molecular mechanisms. However, use of LRS technology is currently limited by high cost, low accessibility, the need for specialised DNA extraction procedures, limited availability of LRS bioinformatic tools and pipelines, and the relative lack of healthy control LRS variant databases. Once these barriers are addressed, the implementation of LRS into clinical diagnostic pipelines will undoubtedly streamline the diagnostic algorithm and increase the diagnostic rate for genetic myopathies. In this review, we discuss the utility and critical impact of LRS in this field.
遗传性肌病由核基因组和线粒体基因组中300多个基因的致病变异引起。尽管短读长下一代测序(NGS)彻底改变了遗传疾病的诊断,但大的和/或复杂的遗传变异在遗传性肌病中占比过高,使用这种方法无法很好地表征。长读长测序(LRS)是一种更新的基因检测技术,克服了NGS的许多局限性。特别是,LRS能更好地检测具有挑战性的变异类型,包括短串联重复序列(STR)扩增、拷贝数变异和结构变异,以及改进变异定相和同时评估表观遗传变化,包括DNA甲基化。鉴于最近描述的与STR扩增相关的遗传性肌病数量不断增加,同时检测多个STR扩增的能力尤为重要。LRS还将有助于鉴定新的肌病基因和分子机制。然而,目前LRS技术的应用受到高成本、低可及性、需要专门的DNA提取程序、LRS生物信息学工具和流程的可用性有限以及相对缺乏健康对照LRS变异数据库的限制。一旦这些障碍得到解决,将LRS应用于临床诊断流程无疑将简化诊断算法并提高遗传性肌病的诊断率。在本综述中,我们讨论了LRS在该领域的实用性和关键影响。