Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia.
Moscow Center for Advanced Studies, Kulakova Str. 20, 123592 Moscow, Russia.
Mar Drugs. 2024 Aug 24;22(9):382. doi: 10.3390/md22090382.
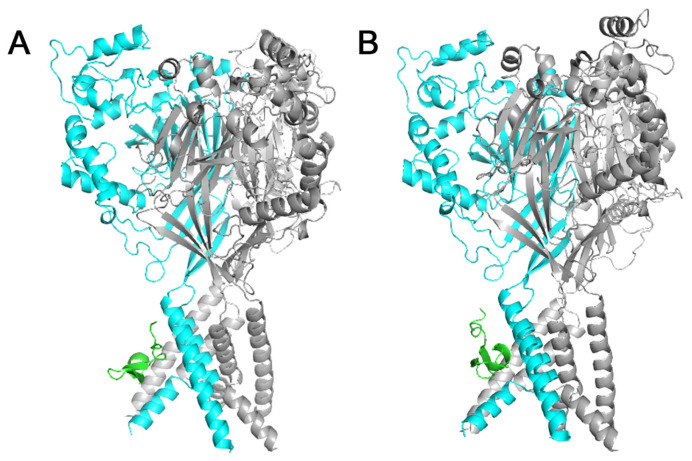
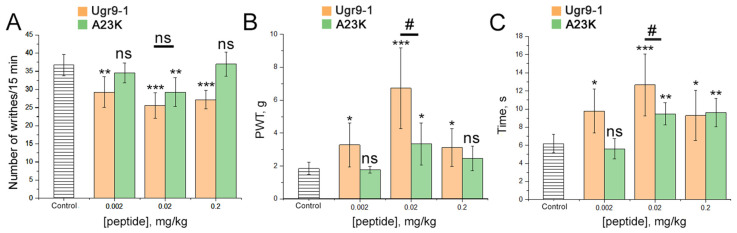
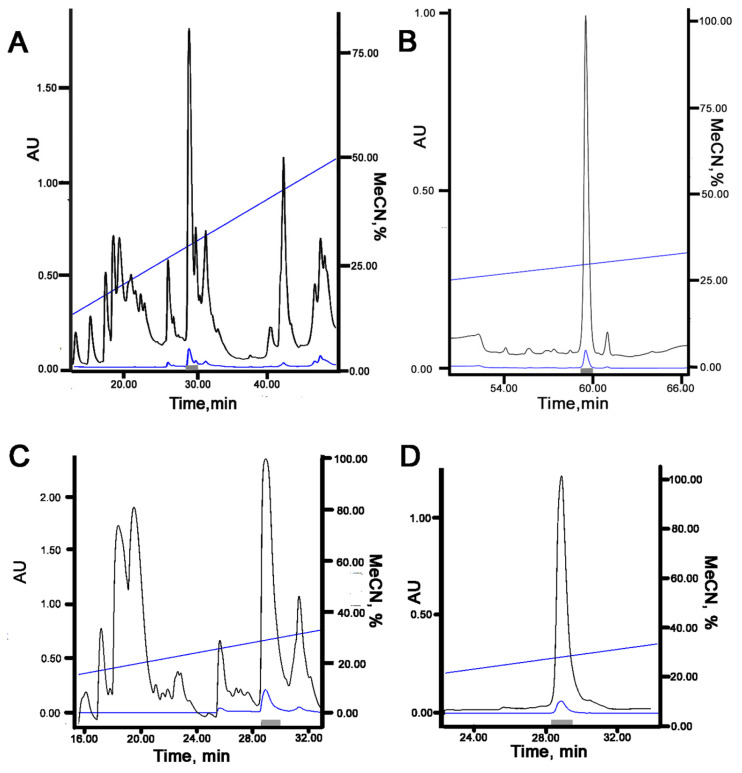
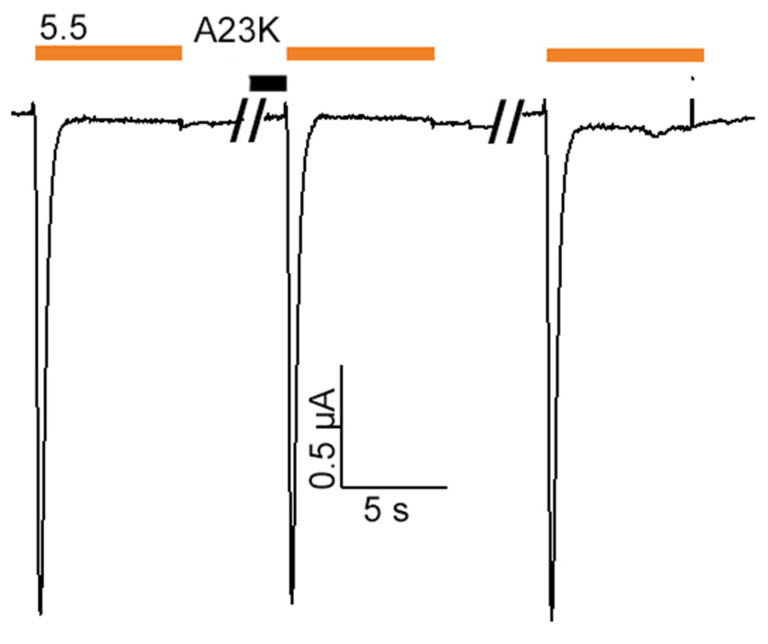
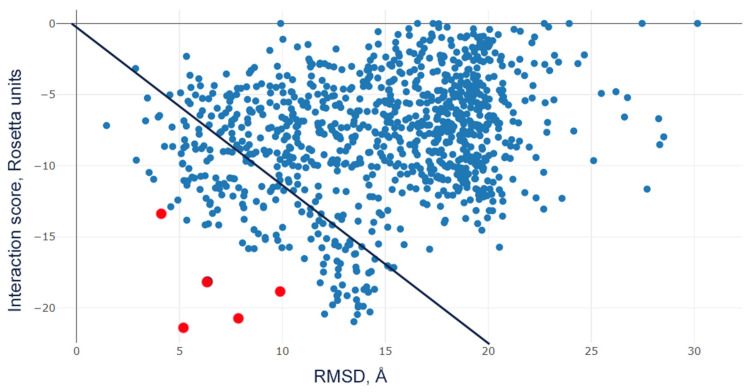
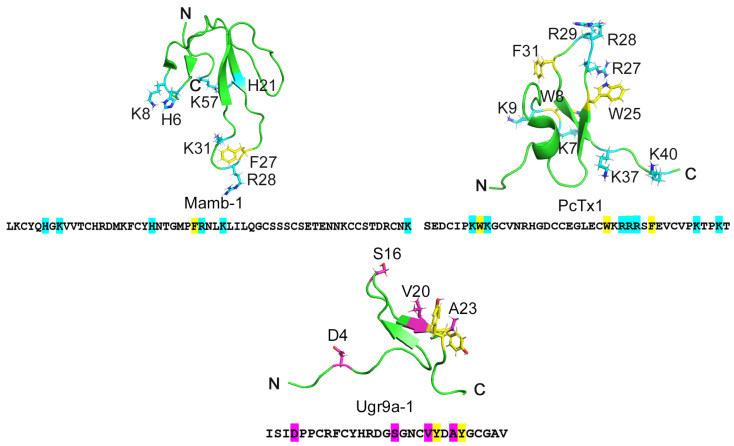
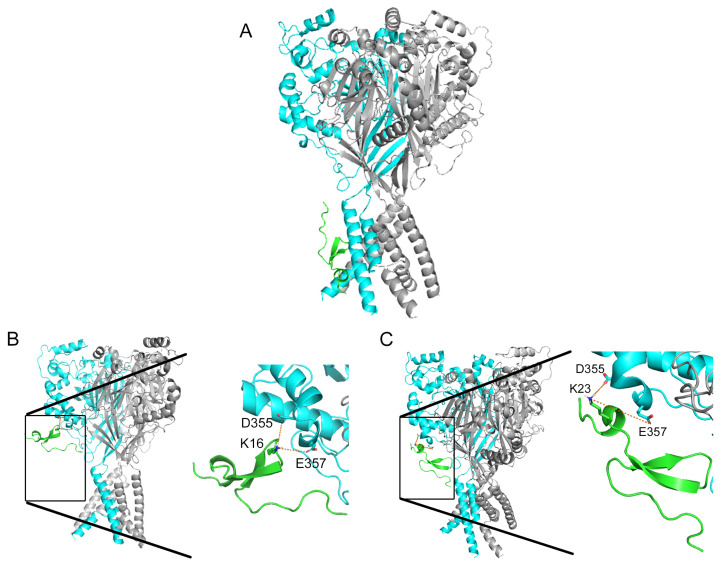
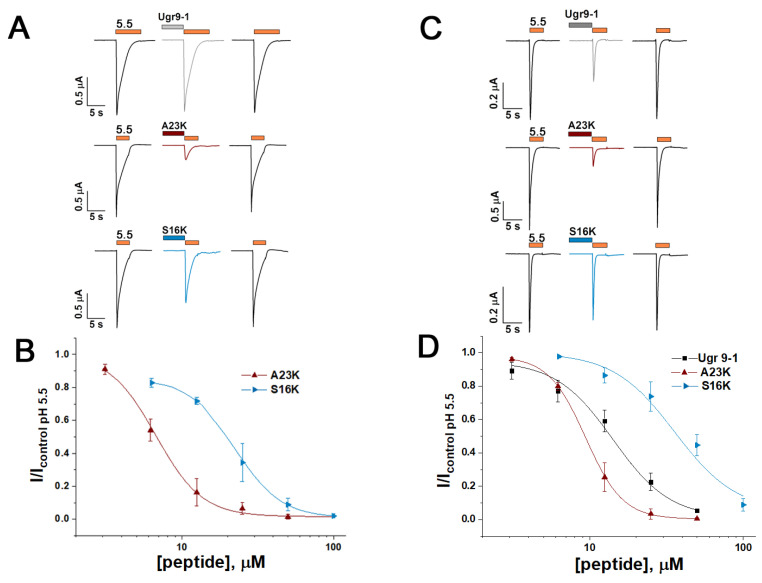
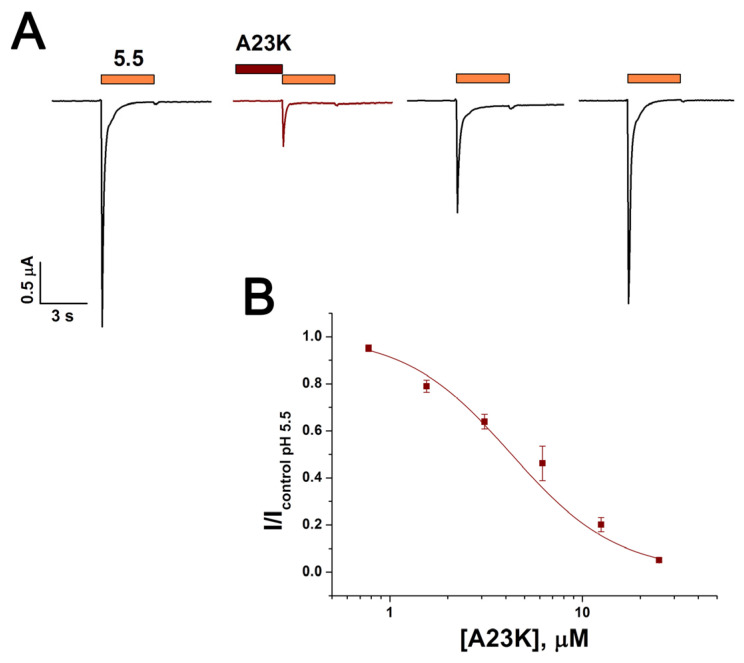
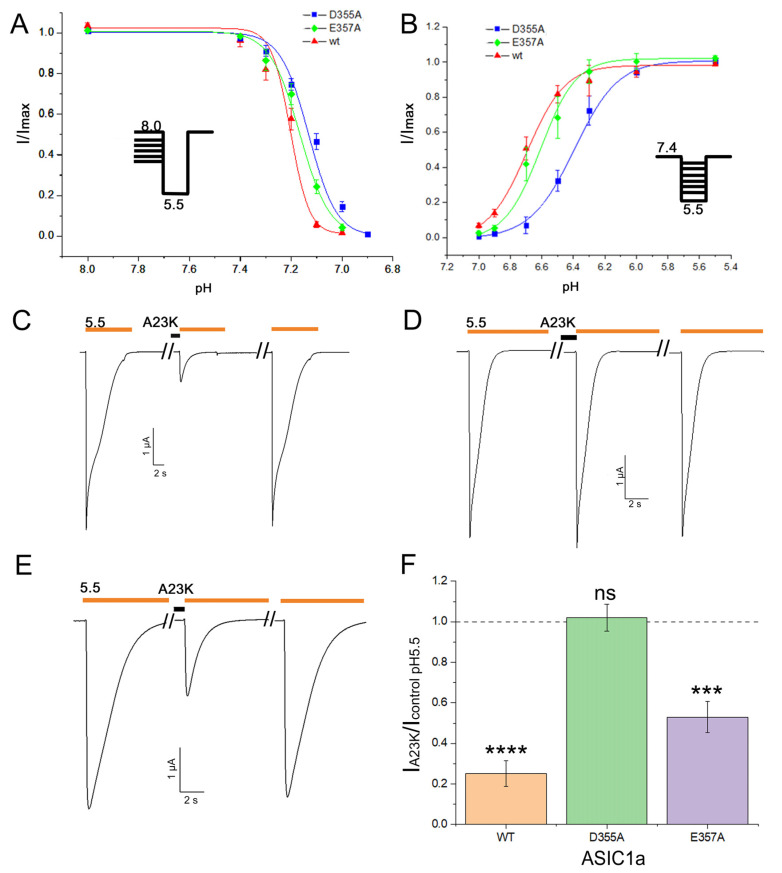
Acid-sensing ion channels (ASICs), which act as proton-gating sodium channels, have garnered attention as pharmacological targets. ASIC1a isoform, notably prevalent in the central nervous system, plays an important role in synaptic plasticity, anxiety, neurodegeneration, etc. In the peripheral nervous system, ASIC1a shares prominence with ASIC3, the latter well established for its involvement in pain signaling, mechanical sensitivity, and inflammatory hyperalgesia. However, the precise contributions of ASIC1a in peripheral functions necessitate thorough investigation. To dissect the specific roles of ASICs, peptide ligands capable of modulating these channels serve as indispensable tools. Employing molecular modeling, we designed the peptide targeting ASIC1a channel from the sea anemone peptide Ugr9-1, originally targeting ASIC3. This peptide (A23K) retained an inhibitory effect on ASIC3 (IC 9.39 µM) and exhibited an additional inhibitory effect on ASIC1a (IC 6.72 µM) in electrophysiological experiments. A crucial interaction between the Lys23 residue of the A23K peptide and the Asp355 residue in the thumb domain of the ASIC1a channel predicted by molecular modeling was confirmed by site-directed mutagenesis of the channel. However, A23K peptide revealed a significant decrease in or loss of analgesic properties when compared to the wild-type Ugr9-1. In summary, using A23K, we show that negative modulation of the ASIC1a channel in the peripheral nervous system can compromise the efficacy of an analgesic drug. These results provide a compelling illustration of the complex balance required when developing peripheral pain treatments targeting ASICs.
酸敏离子通道(ASICs)作为质子门控钠离子通道,作为药理学靶点引起了关注。ASIC1a 同工型在中枢神经系统中尤为普遍,在突触可塑性、焦虑、神经退行性变等方面发挥重要作用。在周围神经系统中,ASIC1a 与 ASIC3 共同突出,后者因其参与疼痛信号传递、机械敏感性和炎症性痛觉过敏而得到充分证实。然而,ASIC1a 在周围功能中的精确贡献需要进行彻底的研究。为了剖析 ASICs 的特定作用,能够调节这些通道的肽配体成为不可或缺的工具。我们采用分子建模,从海葵肽 Ugr9-1 设计靶向 ASIC1a 通道的肽,该肽最初靶向 ASIC3。该肽(A23K)在电生理学实验中保留了对 ASIC3 的抑制作用(IC9.39µM),并对 ASIC1a 表现出额外的抑制作用(IC6.72µM)。分子建模预测的 A23K 肽的 Lys23 残基与 ASIC1a 通道拇指结构域的 Asp355 残基之间的关键相互作用通过通道的定点突变得到了证实。然而,与野生型 Ugr9-1 相比,A23K 肽的镇痛特性显著降低或丧失。总之,使用 A23K,我们表明外周神经系统中 ASIC1a 通道的负向调节可能会影响镇痛药物的疗效。这些结果有力地说明了开发针对 ASICs 的外周疼痛治疗方法时所需的复杂平衡。