Kamp Jan C, Madadi-Sanjani Omid, Uecker Marie, Werlein Christopher, Neubert Lavinia, Kübler Joachim F, Obed Mikal, Junge Norman, Welte Tobias, Ruwisch Jannik, Jonigk Danny D, Stolk Jan, Vieten Gertrud, Janciauskiene Sabina
Department of Respiratory and Infectious Medicine, Hannover Medical School, Hannover, Germany.
Biomedical Research in Endstage and Obstructive Lung Disease Hannover (BREATH), German Center for Lung Research (DZL), Hannover, Germany.
Pediatr Res. 2025 Apr;97(5):1696-1705. doi: 10.1038/s41390-024-03582-w. Epub 2024 Sep 28.
Biliary atresia (BA) is a rare condition of unknown origin in newborns with jaundice. In BA bile ducts are non-functional, causing neonatal cholestasis and following liver fibrosis and failure.
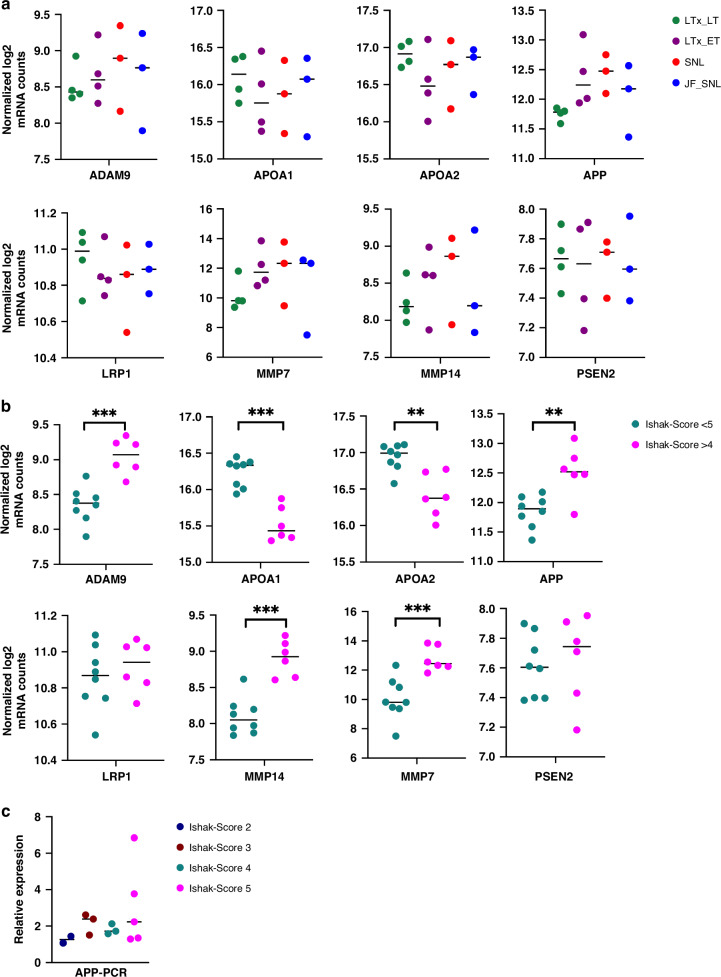
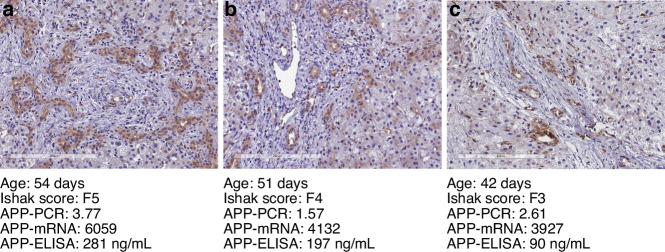
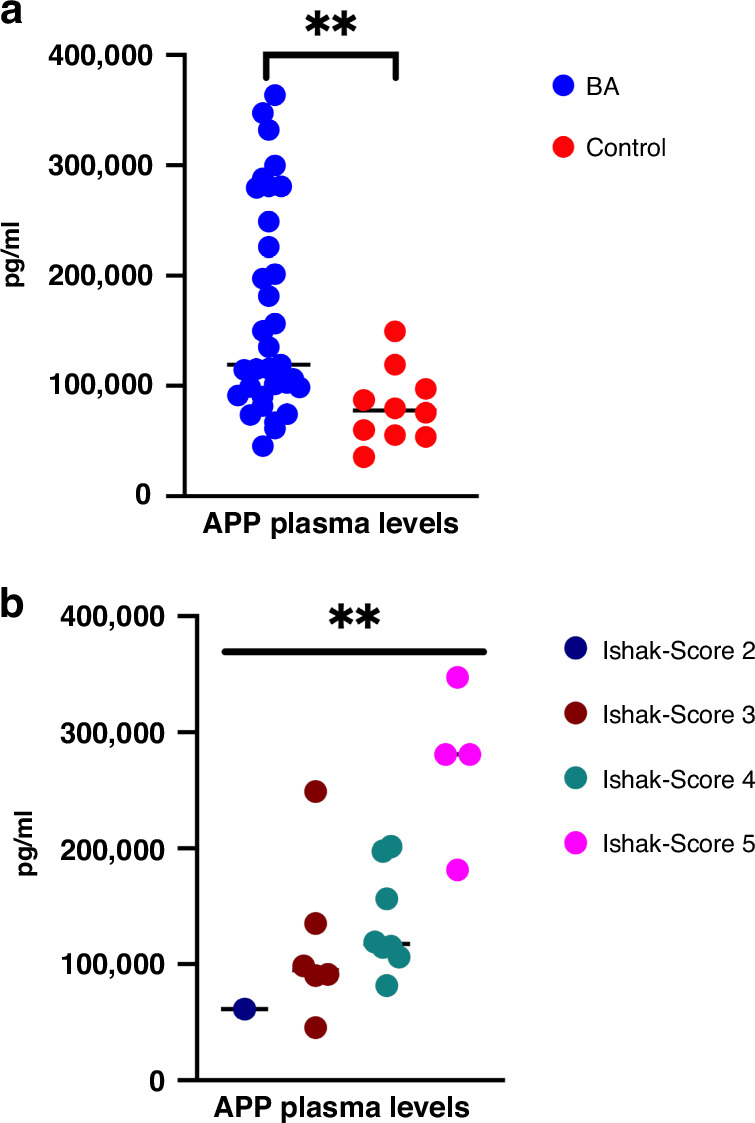
This retrospective study included liver biopsies of 14 infants with BA aged [mean ± SD] 63 ± 23 days. Patients were grouped according to the clinical course (jaundice-free vs recurrent jaundice vs required liver transplantation or liver fibrosis (Ishak fibrosis score)) and followed for 1.61-5.64 years (mean 4.03). Transcriptome profiles were assessed using a panel of 768 fibrosis-specific genes, reanalyzed via qRT-PCR, and confirmed via immunostaining. Plasma from an additional 30 BA infants and 10 age-matched controls were used for amyloid precursor protein (APP) quantification by ELISA.
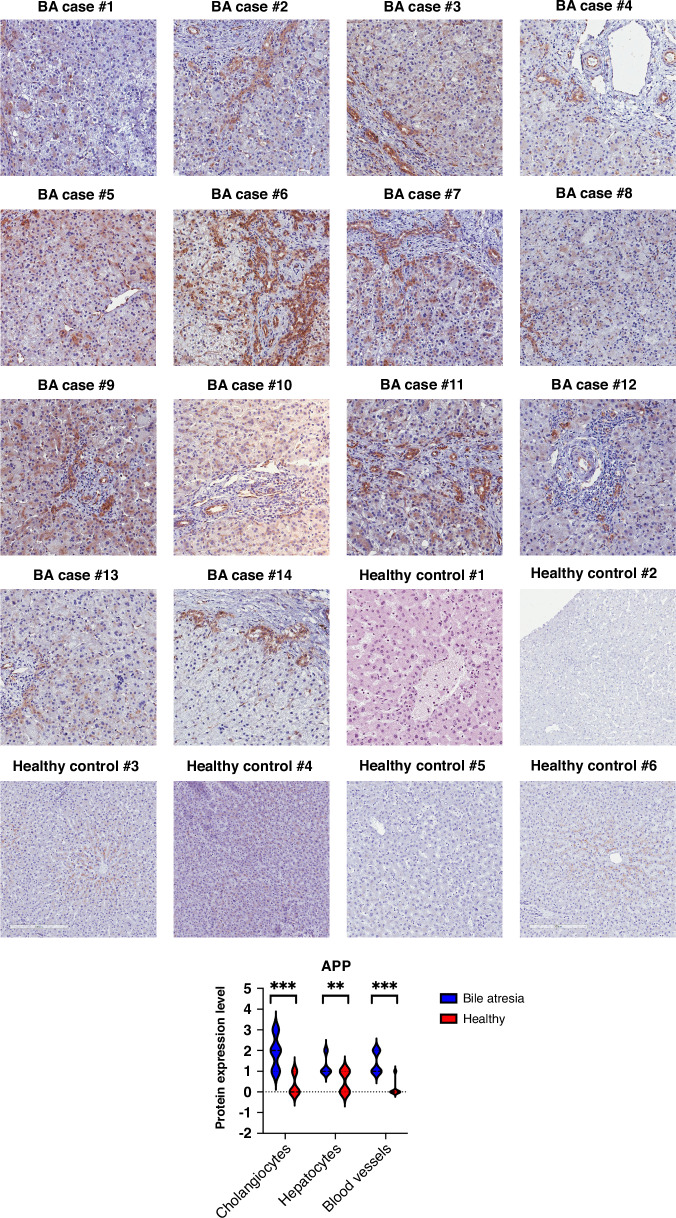
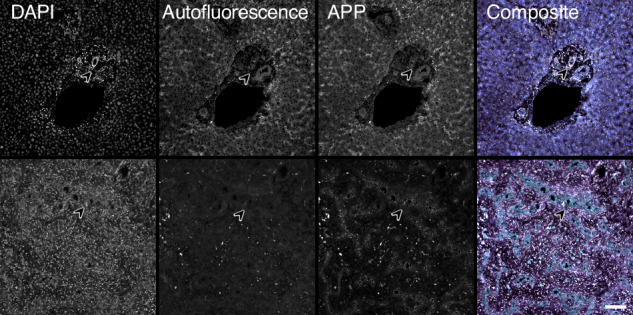
Different clinical outcome groups showed a homogeneous mRNA expression. Altered amyloid-metabolism-related gene expression was found between cases with Ishak fibrosis score greater than 4. Immunostaining confirmed a distinct presence of APP in the livers of all BA subjects. APP plasma levels were higher in BA than in age-matched controls and correlated with the histological fibrosis grade.
These results suggest that amyloidosis may contribute to BA and liver fibrosis, indicating that APP could serve as a potential liquid biomarker for these conditions.
Biliary atresia patients with higher fibrosis scores according to Ishak have higher hepatic expression of amyloid-related genes while amyloid precursor protein accumulates in the liver and increases in the circulation. After a recent study revealed beta-amyloid deposition as a mechanism potentially involved in biliary atresia, we were able to correlate amyloid-metabolism-related transcript levels as well as amyloid precursor protein tissue and plasma levels with the degree of hepatic fibrosis. These findings suggest that amyloid precursor protein is a fibrosis marker in infants with biliary atresia, reinforcing the role of amyloid metabolism in the pathogenesis of this serious disease.
胆道闭锁(BA)是新生儿黄疸中一种病因不明的罕见病症。在BA中,胆管失去功能,导致新生儿胆汁淤积,进而引发肝纤维化和肝功能衰竭。
这项回顾性研究纳入了14例年龄为[均值±标准差]63±23天的BA婴儿的肝活检样本。根据临床病程(无黄疸、复发性黄疸、需要肝移植或肝纤维化(Ishak纤维化评分))对患者进行分组,并随访1.61 - 5.64年(平均4.03年)。使用一组768个纤维化特异性基因评估转录组谱,通过qRT-PCR重新分析,并通过免疫染色进行确认。另外30例BA婴儿和10例年龄匹配的对照的血浆用于通过ELISA定量淀粉样前体蛋白(APP)。
不同临床结局组显示出均匀的mRNA表达。在Ishak纤维化评分大于4的病例之间发现了淀粉样蛋白代谢相关基因表达的改变。免疫染色证实所有BA受试者的肝脏中均有明显的APP存在。BA患者的APP血浆水平高于年龄匹配的对照,且与组织学纤维化分级相关。
这些结果表明淀粉样变性可能导致BA和肝纤维化,这表明APP可能作为这些病症的潜在液体生物标志物。
根据Ishak评分,纤维化评分较高的胆道闭锁患者肝脏中淀粉样相关基因的表达较高,而淀粉样前体蛋白在肝脏中积累并在循环中增加。在最近一项研究揭示β-淀粉样蛋白沉积是可能参与胆道闭锁的一种机制后,我们能够将淀粉样蛋白代谢相关转录水平以及淀粉样前体蛋白组织和血浆水平与肝纤维化程度相关联。这些发现表明淀粉样前体蛋白是胆道闭锁婴儿的纤维化标志物,强化了淀粉样蛋白代谢在这种严重疾病发病机制中的作用。