Kovalenko Elena, Shaheen Layal, Vergasova Ekaterina, Kamelin Alexey, Rubinova Valerya, Kharitonov Dmitry, Kim Anna, Plotnikov Nikolay, Elmuratov Artem, Borovkova Natalia, Storozheva Maya, Solonin Sergey, Gilyazova Irina, Mironov Petr, Khusnutdinova Elza, Petrikov Sergey, Ilinskaya Anna, Ilinsky Valery, Rakitko Alexander
Genotek Ltd., Moscow, Russia.
Genetic Technologies Ltd., Yerevan, Armenia.
Front Med (Lausanne). 2024 Sep 19;11:1409714. doi: 10.3389/fmed.2024.1409714. eCollection 2024.
COVID-19 disease has infected more than 772 million people, leading to 7 million deaths. Although the severe course of COVID-19 can be prevented using appropriate treatments, effective interventions require a thorough research of the genetic factors involved in its pathogenesis.
We conducted a genome-wide association study (GWAS) on 7,124 individuals (comprising 6,400 controls who had mild to moderate COVID-19 and 724 cases with severe COVID-19). The inclusion criteria were acute respiratory distress syndrome (ARDS), acute respiratory failure (ARF) requiring respiratory support, or CT scans indicative of severe COVID-19 infection without any competing diseases. We also developed a polygenic risk score (PRS) model to identify individuals at high risk.
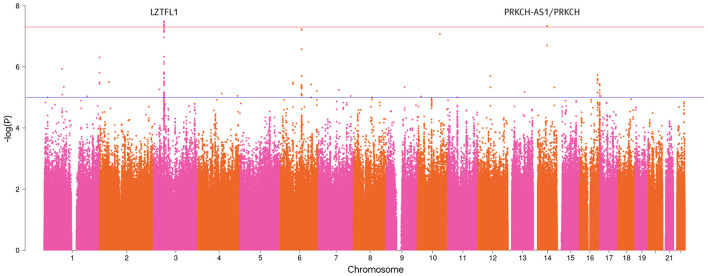
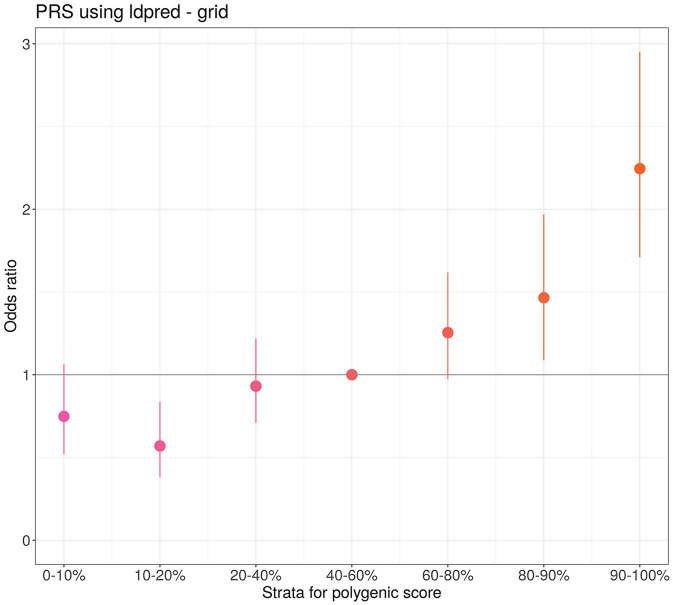
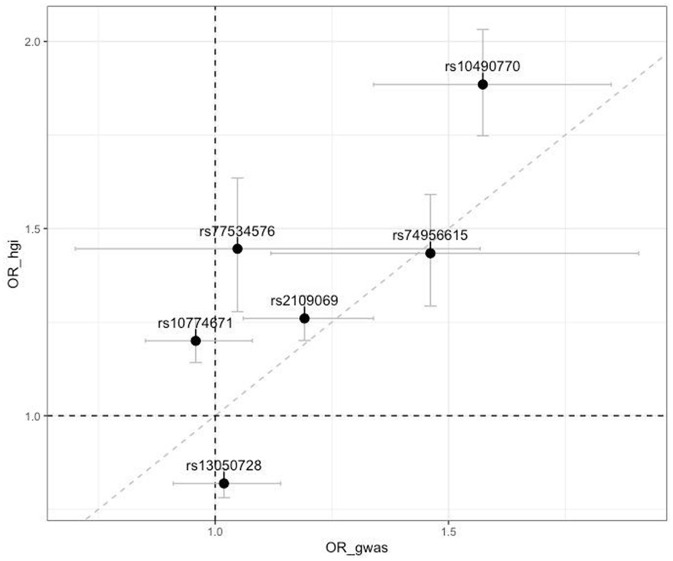
We identified two genome-wide significant loci (-value <5 × 10) and one locus with approximately genome-wide significance (-value = 5.92 × 10-6.15 × 10). The most genome-wide significant variants were located in the () gene, which has been highlighted in several previous GWAS studies. Our PRS model results indicated that individuals in the top 10% group of the PRS had twice the risk of severe course of the disease compared to those at median risk [odds ratio = 2.18 (1.66, 2.86), -value = 8.9 × 10].
We conducted one of the largest studies to date on the genetics of severe COVID-19 in an Eastern European cohort. Our results are consistent with previous research and will guide further epidemiologic studies on host genetics, as well as for the development of targeted treatments.
新冠病毒病已感染超过7.72亿人,导致700万人死亡。尽管使用适当的治疗方法可以预防新冠病毒病的严重病程,但有效的干预措施需要对其发病机制中涉及的遗传因素进行深入研究。
我们对7124名个体进行了全基因组关联研究(GWAS)(包括6400名患有轻至中度新冠病毒病的对照者和724名患有严重新冠病毒病的病例)。纳入标准为急性呼吸窘迫综合征(ARDS)、需要呼吸支持的急性呼吸衰竭(ARF)或CT扫描显示为严重新冠病毒感染且无任何并发疾病。我们还开发了一种多基因风险评分(PRS)模型来识别高危个体。
我们确定了两个全基因组显著位点(P值<5×10⁻⁸)和一个具有近似全基因组显著性的位点(P值 = 5.92×10⁻⁶至6.15×10⁻⁶)。最具全基因组显著性的变异位于()基因中,该基因在之前的多项GWAS研究中已被重点关注。我们的PRS模型结果表明,PRS排名前10%的个体发生严重病程的风险是中等风险个体的两倍[优势比 = 2.18(1.66,2.86),P值 = 8.9×10⁻⁸]。
我们在东欧队列中进行了迄今为止关于严重新冠病毒病遗传学的最大规模研究之一。我们的结果与先前的研究一致,将指导进一步关于宿主遗传学的流行病学研究以及靶向治疗的开发。