National Centre for Animal Disease, Canadian Food Inspection Agency, Lethbridge County, AB, T1J 5R7, Canada.
Saskatoon Research and Development Centre, Agriculture and Agri-Food Canada, Saskatoon, SK, S7N 0X2, Canada.
Bioinformatics. 2024 Oct 1;40(10). doi: 10.1093/bioinformatics/btae601.
State-of-the-art tools for classifying metagenomic sequencing reads provide both rapid and accurate options, although the combination of both in a single tool is a constantly improving area of research. The machine learning-based Naïve Bayes Classifier (NBC) approach provides a theoretical basis for accurate classification of all reads in a sample.
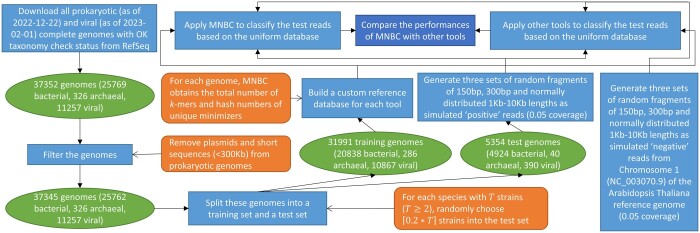
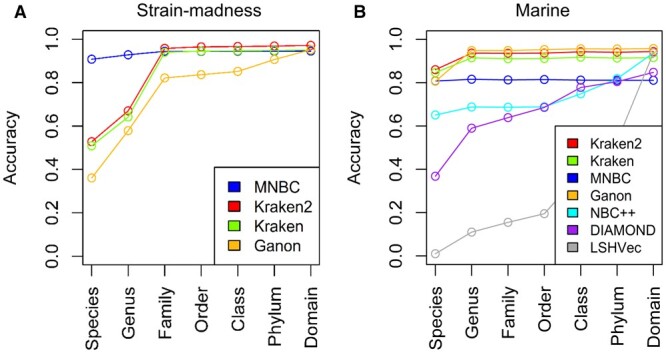
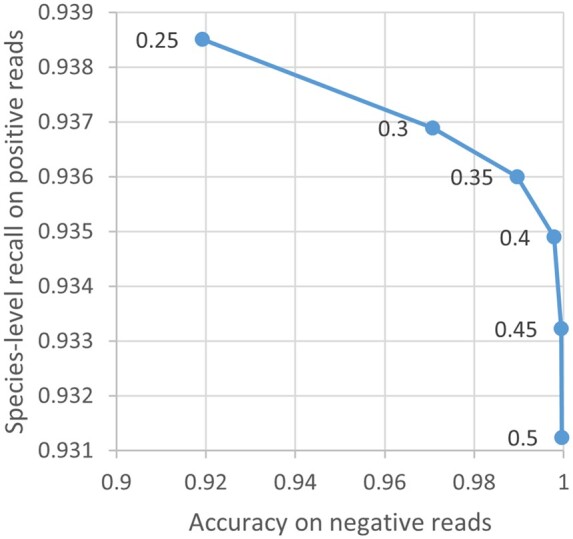
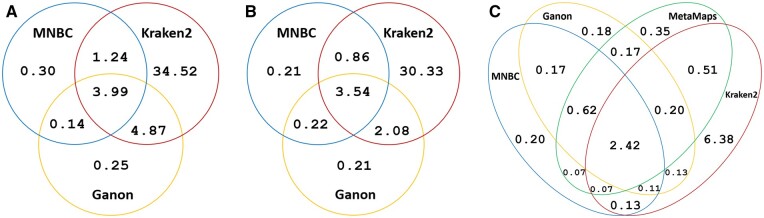
We developed the multithreaded Minimizer-based Naïve Bayes Classifier (MNBC) tool to improve the NBC approach by applying minimizers, as well as plurality voting for closely related classification scores. A standard reference- and test-sequence framework using simulated variable-length reads benchmarked MNBC with six other state-of-the-art tools: MetaMaps, Ganon, Kraken2, KrakenUniq, CLARK, and Centrifuge. We also applied MNBC to the "marine" and "strain-madness" short-read metagenomic datasets in the Critical Assessment of Metagenome Interpretation (CAMI) II challenge using a corresponding database from the time. MNBC efficiently identified reads from unknown microorganisms, and exhibited the highest species- and genus-level precision and recall on short reads, as well as the highest species-level precision on long reads. It also achieved the highest accuracy on the "strain-madness" dataset.
MNBC is freely available at: https://github.com/ComputationalPathogens/MNBC.
用于分类宏基因组测序reads 的最先进工具提供了快速且准确的选项,尽管将这两者组合在一个工具中是一个不断改进的研究领域。基于机器学习的朴素贝叶斯分类器(NBC)方法为准确分类样本中的所有reads 提供了理论基础。
我们开发了基于多线程 Minimizer 的朴素贝叶斯分类器(MNBC)工具,通过应用 minimizers 以及对密切相关的分类分数进行多数投票,改进了 NBC 方法。使用模拟可变长度 reads 的标准参考和测试序列框架,使用六个其他最先进的工具对 MNBC 进行了基准测试:MetaMaps、Ganon、Kraken2、KrakenUniq、CLARK 和 Centrifuge。我们还使用相应的数据库,将 MNBC 应用于 Critical Assessment of Metagenome Interpretation (CAMI) II 挑战中的“海洋”和“菌株疯狂”短读宏基因组数据集。MNBC 能够有效地识别未知微生物的reads,并在短reads 上表现出最高的物种和属水平的精度和召回率,在长reads 上表现出最高的物种水平的精度,在“菌株疯狂”数据集上也达到了最高的准确性。
MNBC 可在以下网址免费获取:https://github.com/ComputationalPathogens/MNBC。