Tisch Adam, Madapoosi Siddharth, Blough Stephen, Rosa Jan, Eddy Sean, Mariani Laura, Naik Abhijit, Limonte Christine, McCown Philip, Menon Rajasree, Rosas Sylvia E, Parikh Chirag R, Kretzler Matthias, Mahfouz Ahmed, Alakwaa Fadhl
Undergraduate Research Opportunity Program, University of Michigan, Ann Arbor, MI, USA.
University of Michigan Medical School, Ann Arbor, MI, USA.
Heliyon. 2024 Sep 27;10(19):e38567. doi: 10.1016/j.heliyon.2024.e38567. eCollection 2024 Oct 15.
Single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA sequencing (snRNA-seq) provide valuable insights into the cellular states of kidney cells. However, the annotation of cell types often requires extensive domain expertise and time-consuming manual curation, limiting scalability and generalizability. To facilitate this process, we tested the performance of five supervised classification methods for automatic cell type annotation.
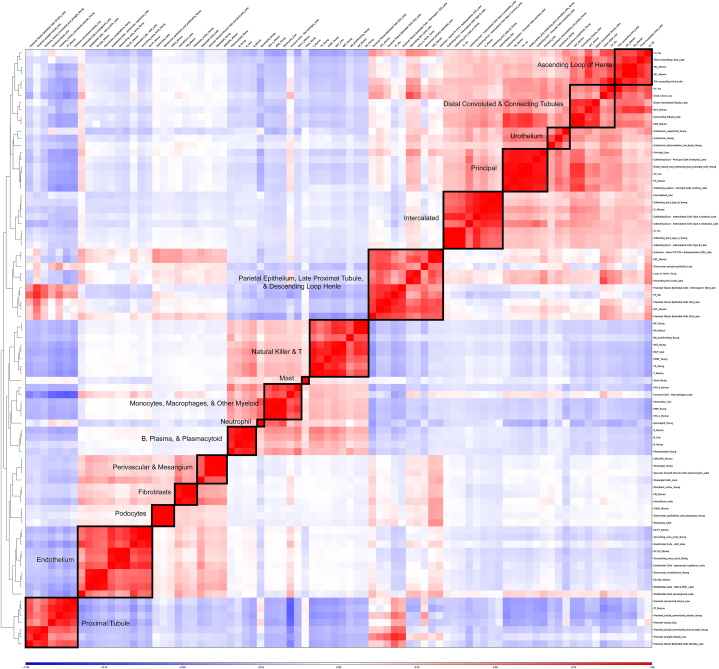
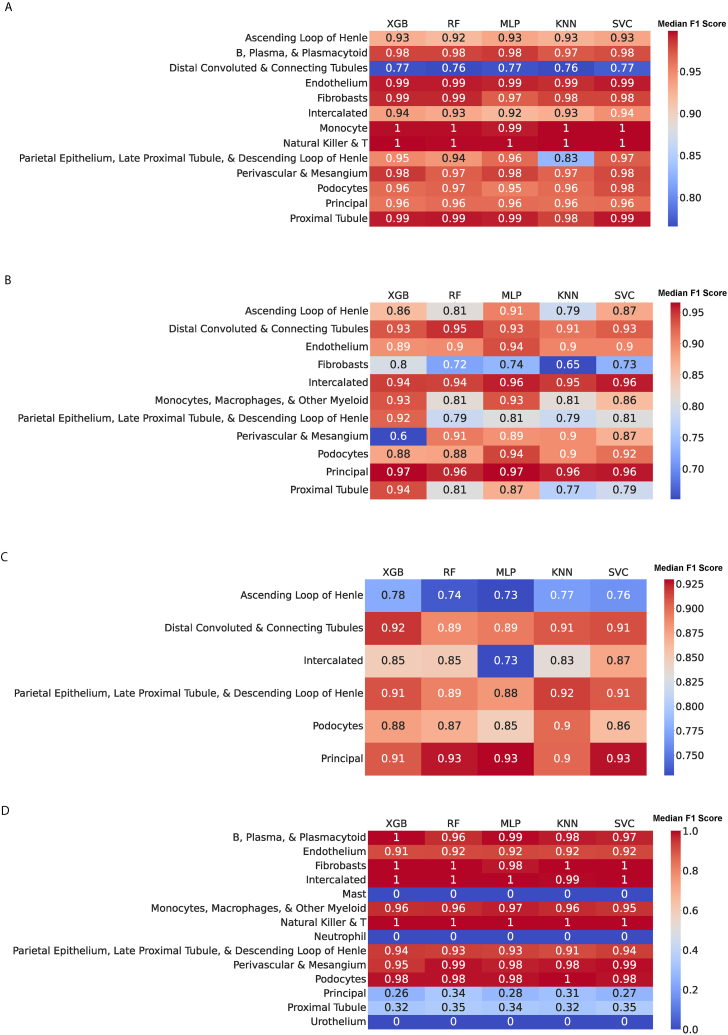
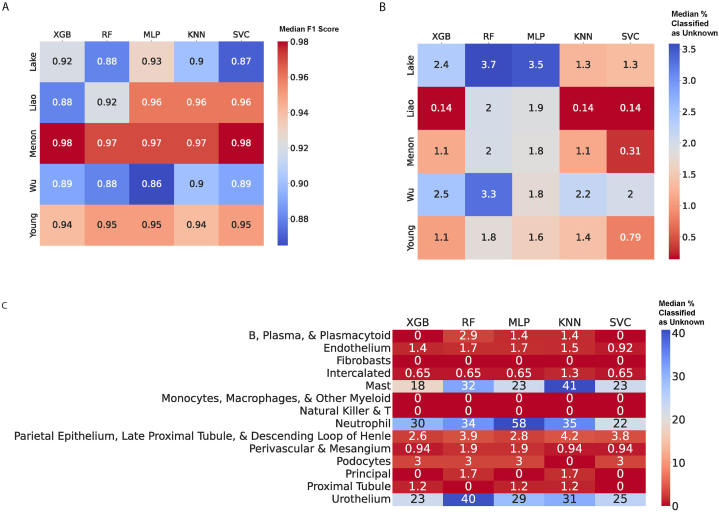
We analyzed publicly available sc/snRNA-seq datasets from five expert-annotated studies, comprising 62,120 cells from 79 kidney biopsy samples. Datasets were integrated by harmonizing cell type annotations across studies. Five different supervised machine learning algorithms (support vector machines, random forests, multilayer perceptrons, k-nearest neighbors, and extreme gradient boosting) were applied to automatically annotate cell types using four training datasets and one testing dataset. Performance metrics, including accuracy (F1 score) and rejection rates, were evaluated. All five machine learning algorithms demonstrated high accuracies, with a median F1 score of 0.94 and a median rejection rate of 1.8 %. The algorithms performed equally well across different datasets and successfully rejected cell types that were not present in the training data. However, F1 scores were lower when models trained primarily on scRNA-seq data were tested on snRNA-seq data.
Despite limitations including the number of biopsy samples, our findings demonstrate that machine learning algorithms can accurately annotate a wide range of adult kidney cell types in scRNA-seq/snRNA-seq data. This approach has the potential to standardize cell type annotation and facilitate further research on cellular mechanisms underlying kidney disease.
单细胞RNA测序(scRNA-seq)和单细胞核RNA测序(snRNA-seq)为了解肾细胞的细胞状态提供了有价值的见解。然而,细胞类型的注释通常需要广泛的领域专业知识和耗时的人工整理,这限制了可扩展性和通用性。为了促进这一过程,我们测试了五种监督分类方法用于自动细胞类型注释的性能。
我们分析了来自五项经过专家注释研究的公开可用sc/snRNA-seq数据集,包括来自79份肾活检样本的62,120个细胞。通过协调各研究中的细胞类型注释来整合数据集。使用四个训练数据集和一个测试数据集,应用五种不同的监督机器学习算法(支持向量机、随机森林、多层感知器、k近邻和极端梯度提升)自动注释细胞类型。评估了包括准确率(F1分数)和拒绝率在内的性能指标。所有五种机器学习算法都表现出很高的准确率,F1分数中位数为0.94,拒绝率中位数为1.8%。这些算法在不同数据集上表现同样出色,并成功拒绝了训练数据中不存在的细胞类型。然而,当主要在scRNA-seq数据上训练的模型在snRNA-seq数据上进行测试时,F1分数较低。
尽管存在包括活检样本数量在内的局限性,但我们的研究结果表明,机器学习算法可以准确注释scRNA-seq/snRNA-seq数据中广泛的成年肾细胞类型。这种方法有可能使细胞类型注释标准化,并促进对肾脏疾病潜在细胞机制的进一步研究。