Fiedler Alexandra, Brittan Kevin, Manatsathit Wuttiporn
Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE.
Division of Gastroenerology and Hepatology, University of Creighton, Omaha, NE.
ACG Case Rep J. 2024 Oct 12;11(10):e01537. doi: 10.14309/crj.0000000000001537. eCollection 2024 Oct.
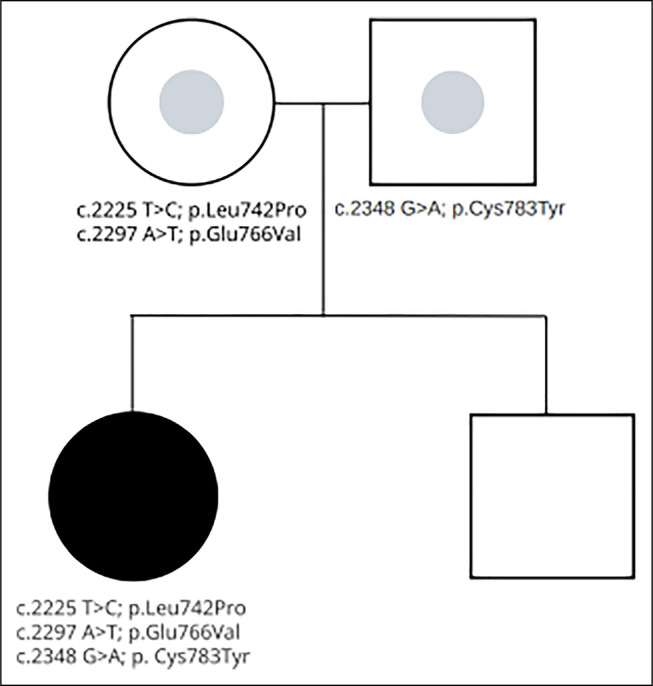
Syndromes characterized by congenital diarrhea, hearing loss, and intrahepatic cholestasis are uncommon and often misdiagnosed as progressive familial intrahepatic cholestasis (PFIC). Recent genetic studies have widened the array of genes linked with cholestatic disorders. Among these, has recently been implicated in microvillous inclusion disease (MVID), although only a few cases exist. This case highlights a 20-year-old woman initially diagnosed clinically with PFIC type 1 during childhood. After ileal bypass at age 4 years, she had a resolution of intractable pruritus and cholestasis. Despite remaining symptom-free for over a decade, she returned in adulthood with recurrent cholestatic pruritus. Odevixibat was initiated for presumed PFIC while awaiting additional testing with symptomatic improvement and laboratory normalization. Whole genome sequencing identified novel compound heterozygous mutations in and small bowel biopsies confirmed villous atrophy. Odevixibat, currently approved for cholestatic pruritus in PFIC and Alagille syndrome, demonstrates efficacy in managing cholestatic pruritus in MVID.
以先天性腹泻、听力丧失和肝内胆汁淤积为特征的综合征并不常见,常被误诊为进行性家族性肝内胆汁淤积症(PFIC)。最近的基因研究扩大了与胆汁淤积性疾病相关的基因范围。其中, 最近被认为与微绒毛包涵体病(MVID)有关,尽管仅有少数病例。本病例突出了一名20岁女性,她在儿童期最初临床诊断为1型PFIC。4岁时进行回肠转流术后,她顽固性瘙痒和胆汁淤积症状得到缓解。尽管十多年来一直无症状,但成年后她因复发性胆汁淤积性瘙痒再次就诊。在等待进一步检查期间,因推测为PFIC开始使用odevixibat治疗,症状有所改善,实验室指标恢复正常。全基因组测序在 中发现了新的复合杂合突变,小肠活检证实存在绒毛萎缩。Odevixibat目前已被批准用于治疗PFIC和阿拉吉尔综合征的胆汁淤积性瘙痒,在治疗MVID的胆汁淤积性瘙痒方面显示出疗效。