Saadabadi Atefeh, Rantanen Marja, Marimuthu Parthiban, Koivisto Ari-Pekka, Eklund Patrik C, Salo-Ahen Outi M H
Pharmaceutical Sciences Laboratory, Faculty of Science and Engineering, Åbo Akademi University, Tykistökatu 6, Turku, 20520, Finland.
Laboratory of Molecular Science and Engineering, Faculty of Science and Engineering, Åbo Akademi University, Henrikinkatu 2, Turku, 20500, Finland.
ChemMedChem. 2025 Feb 1;20(3):e202400501. doi: 10.1002/cmdc.202400501. Epub 2024 Nov 20.
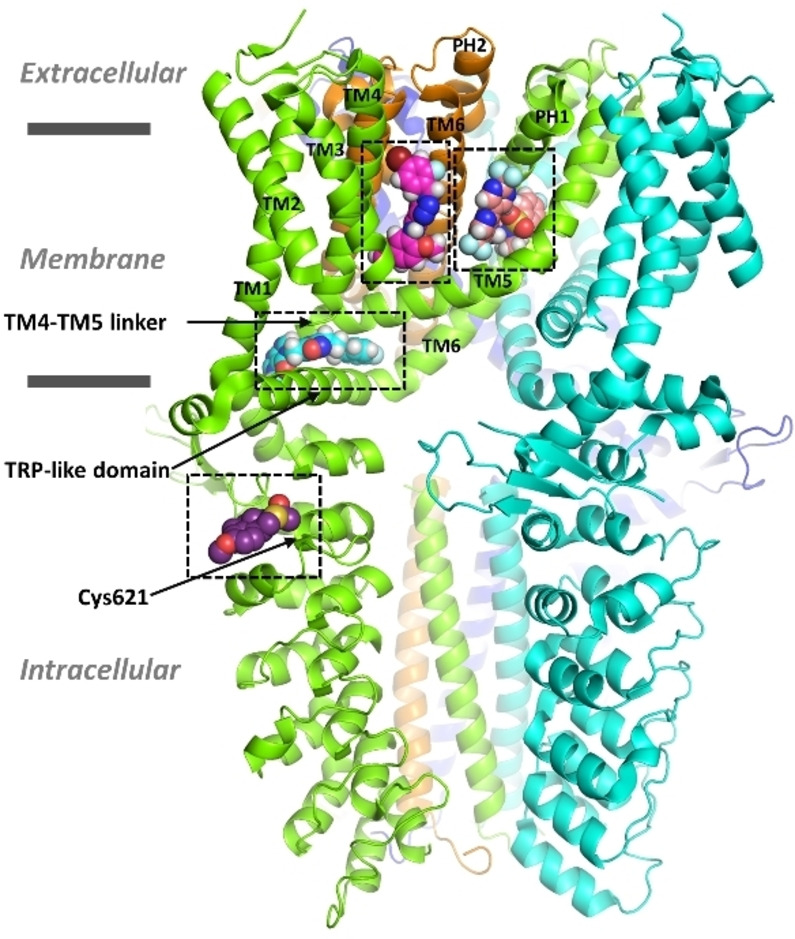
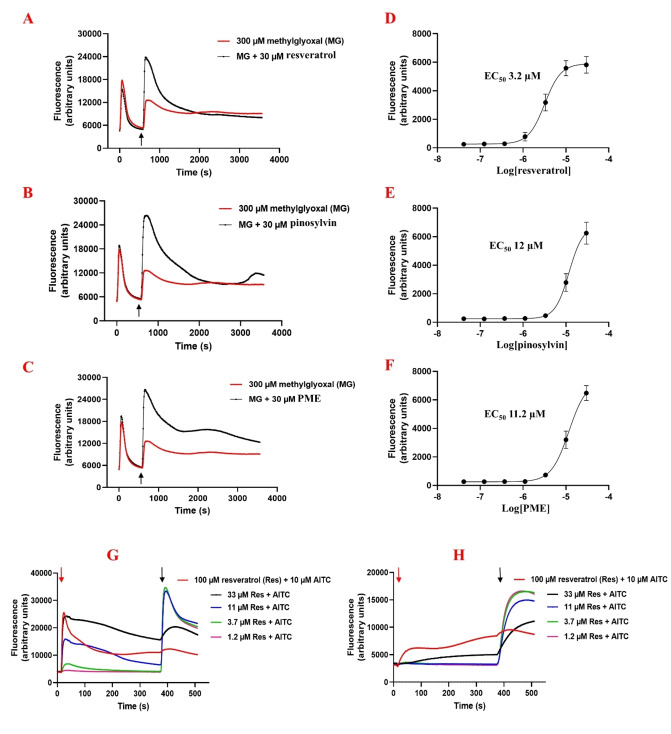
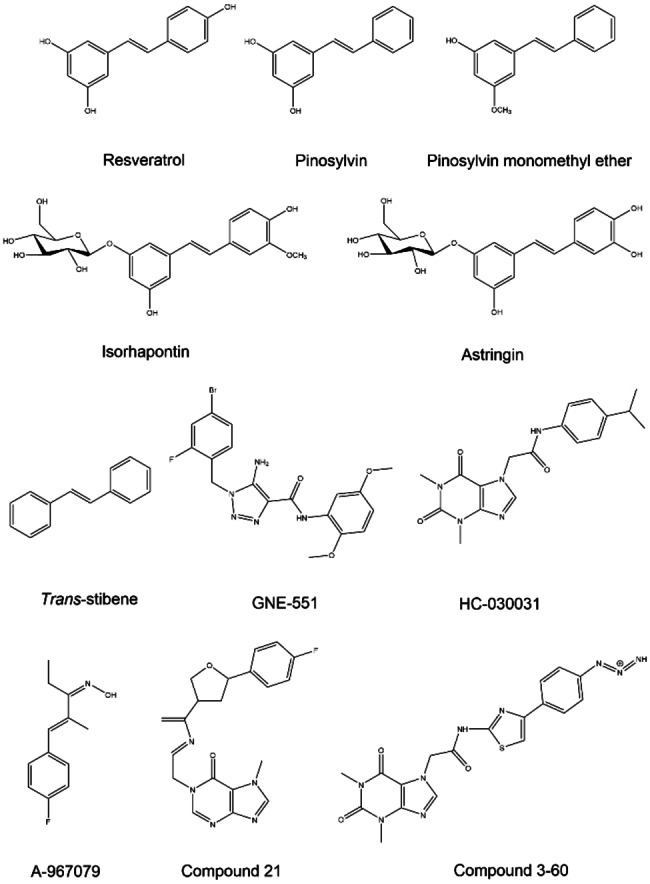
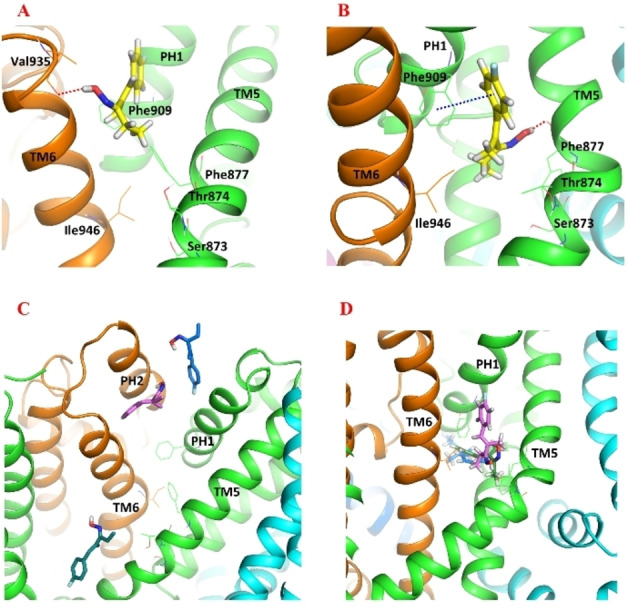
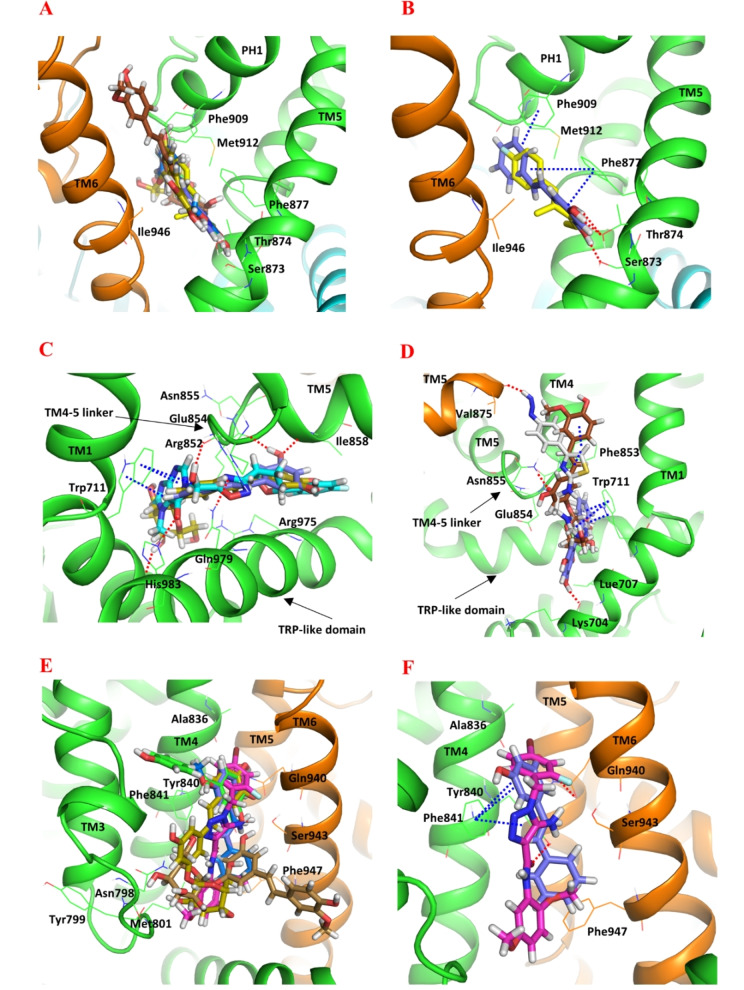
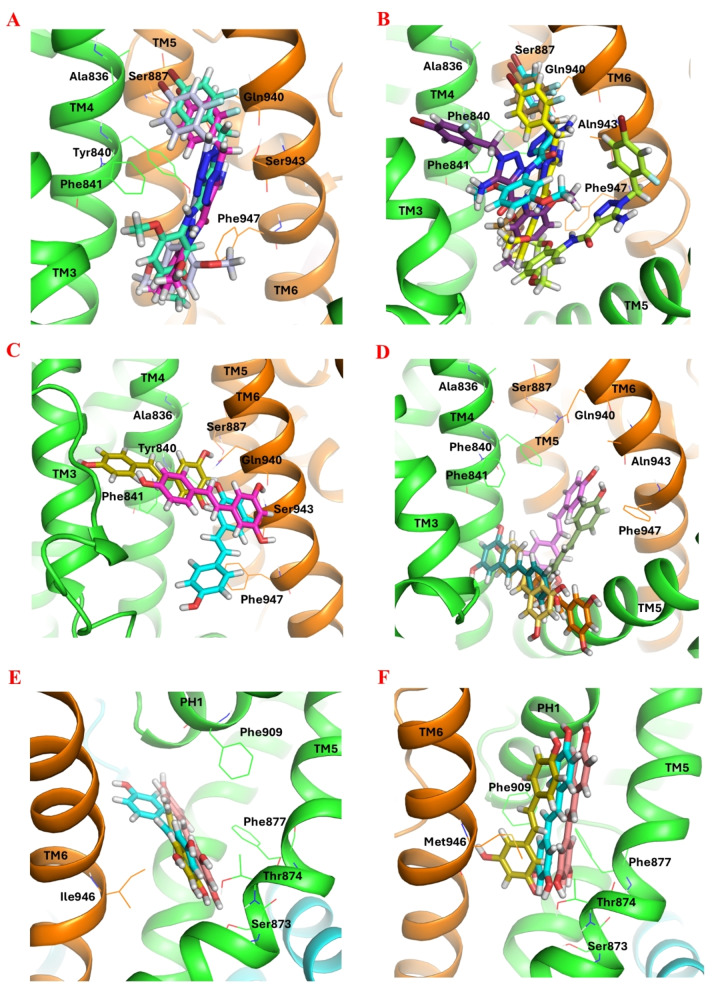
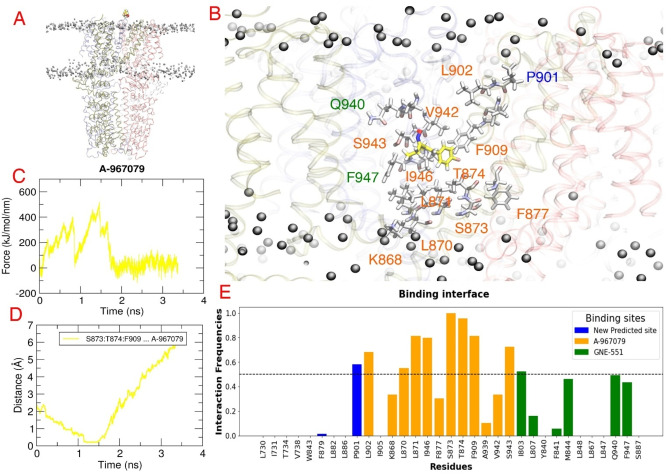
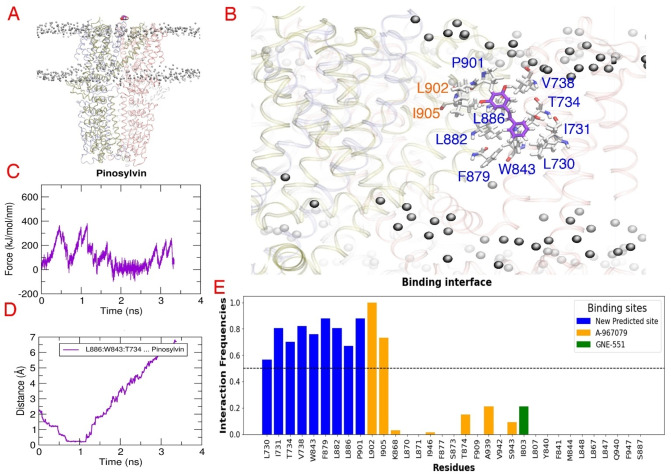
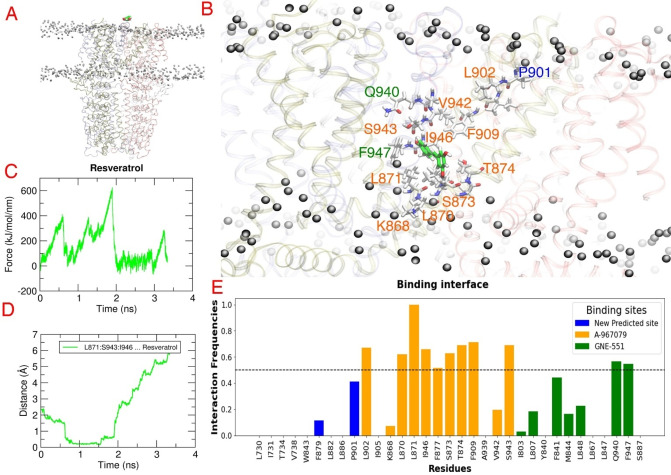
Natural stilbenoids, polyphenolic compounds notably found in Scots pine and Norway spruce, have been shown to exhibit analgesic and anti-inflammatory effects through the TRPA1 channel, making them promising hits for the development of novel agents to treat inflammatory diseases and pain. In this study, we computationally investigated the putative binding sites of natural stilbenoids at the TRPA1 channel. Specifically, we employed molecular docking and MD simulation approaches to explore three known ligand binding sites at TRPA1. Furthermore, the biological effect of the studied compounds on TRPA1 was assessed in vitro using a fluorescent imaging plate reader (FLIPR™) calcium assay. Our modeling results suggest the stilbenoids exhibit higher affinity to the two agonist binding sites than the antagonistic site. Consistent with this, the in vitro results showed that the stilbenoids act as moderate TRPA1 channel agonists and likely inhibit the channel through a desensitization mechanism rather than act as pure TRPA1 antagonists. Additionally, our bias-force pulling simulations proposed an additional binding pocket for the natural stilbenoids that is distinct from the known ligand binding sites at TRPA1. The results of the study give useful insights into structure-based design and development of novel therapeutic TRPA1 modulators.
天然芪类化合物是在欧洲赤松和挪威云杉中特别发现的多酚类化合物,已被证明可通过TRPA1通道发挥镇痛和抗炎作用,使其成为开发治疗炎症性疾病和疼痛的新型药物的有前景的候选物。在本研究中,我们通过计算研究了天然芪类化合物在TRPA1通道上的假定结合位点。具体而言,我们采用分子对接和分子动力学模拟方法来探索TRPA1上三个已知的配体结合位点。此外,使用荧光成像微孔板读数仪(FLIPR™)钙测定法在体外评估了所研究化合物对TRPA1的生物学效应。我们的建模结果表明,芪类化合物对两个激动剂结合位点的亲和力高于拮抗剂结合位点。与此一致的是,体外结果表明芪类化合物作为中度TRPA1通道激动剂,可能通过脱敏机制抑制该通道,而不是作为纯TRPA1拮抗剂。此外,我们的偏置力拉动模拟提出了一个天然芪类化合物的额外结合口袋,它与TRPA1上已知的配体结合位点不同。该研究结果为基于结构的新型治疗性TRPA1调节剂的设计和开发提供了有用的见解。